Historical and Current Data Inform the Ovarian Treatment Landscape for Practicing Oncologists
Gynecologic oncology relies on both historical and modern therapies for the treatment of patients with ovarian cancer. During a Targeted Oncology Case-Based Peer Perspective virtual event, Michael J. Birrer, MD, PhD, reviewed the data.


Michael J. Birrer, MD, PhD

Gynecologic oncology relies on both historical and modern therapies for the treatment of patients with ovarian cancer. During a Targeted Oncology Case-Based Peer Perspective virtual event, Michael J. Birrer, MD, PhD, the vice chancellor director of the Winthrop P. Rockefeller CancerInstitute at the University of Arkansas for Medical Sciences in Little Rock, AR, reviewed the data.
Targeted OncologyTM: How do some of the historical data regarding ovarian cancer still play a role in this setting?
BIRRER: With antiangiogenic agents and, in particular, the phase 3 trial GOG-0218 [NCT00262847; investigators] looked at the potential role of bevacizumab [Avastin] in the treatment of patients with advanced-stage ovarian cancer.1 This was a double-blind, randomized, controlled, phase 3 trial, which is fairly large. It had about 1900 women with stage III and IV disease and all of them received chemotherapy. Patients were randomized to 1 of 3 arms; the control arm was carboplatin at area under the curve of 6, paclitaxel at 175 mg/m2. It was placebo controlled.
The first experimental arm used the same chemotherapy backbone but added in concurrent bevacizumab at 15 mg/kg during the chemotherapy, and there was a placebo control on the maintenance part of it. The other experimental arm included bevacizumab throughout, both concurrently and then in the maintenance phase for 15 months or 22 cycles.
The primary end point was PFS [progression-free survival] and [concerning] the baseline characteristics—[I emphasize] that fairly sick population—40% were suboptimally debulked and 26% were stage IV.
The primary end point for the control arm showed a median PFS of 12 months.2 The concurrent bevacizumab treatment arm was essentially indistinguishable at 12.8 months, but the experimental arm that included concurrent bevacizumab and maintenance bevacizumab was 18.2 months. This had a hazard ratio of approximately 0.62 [95% CI, 0.52-0.75; P < .0001], which was highly statistically significant.
What’s important to note is that there was no survival difference in this study [at] 103-month follow-up. You can’t separate the Kaplan-Meier curves. It had such long follow-up that the crossover...was quite large; so that, I think, muddies the potential overall survival [OS] effect of bevacizumab.
The OS and PFS by prognostic biomarker...[showed that] the genetic makeup of the group didn’t really make a difference. The OS difference showed an advantage to patients having either homologous repair deficiency [HRD] or BRCA mutations. We know that because those patients do better prognostically.
The adverse events [AEs] associated with bevacizumab [weren’t] shocking. The important point to look at is the control arm versus the bevacizumab plus maintenance arm; hypertension stands out at 7.2% versus 23.1%, [respectively], for grade 2 or greater. I’ve never had a patient come off bevacizumab for hypertension; [it’s] usually controllable.
The gastrointestinal [GI] events were something that people were concerned about because in the early phase 2 trial, they had several bowel perforations, including deaths. It turns out that in patients [with new diagnoses], while there is a statistically significant increase in GI events, it’s modest and it almost always occurs in the first cycle. There were some thromboembolic events and bleeding, and I always describe that to my patients.
Bevacizumab is one of the options for maintenance therapy and in frontline therapy for patients with ovarian cancer.
What are some newer therapies for these patients, and how do the trials compare for them?
The relatively new kids on the block are PARP inhibitors... for primary maintenance in ovarian cancer. The SOLO-1 trial [NCT01844986] was notable [because] it focused on patients with BRCA mutations.3 The setting was in maintenance. It was a placebo-controlled study and then the initial result was that median PFS was not reached with olaparib [Lynparza] versus 13.8 months with placebo.
The PRIMA trial [NCT02655016]...led to an FDA approval.4 The difference between PRIMA and SOLO-1, other than some differences in the patient population, was that [PRIMA] was for all-comers and it was niraparib [Zejula] versus placebo. The results [for all comers] with PRIMA [showed a] PFS hazard ratio of 0.62 [95% CI, 0.50-0.76]. If you look at the BRCA-mutated patient population, [the PFS hazard ratio was] 0.43, a little different from 0.30 [with SOLO-1]3 but still quite spectacular.
Then PAOLA-1 [NCT02477644], I think, is an interesting study. It’s similar to PRIMA in that it was with all comers and was maintenance, but the difference is that it compared [olaparib] plus bevacizumab versus bevacizumab plus placebo.5 The comparator arm was a treatment arm. It had maintenance therapy, whereas [the other 2 were] just placebo. The [all comer population had a PFS of] 22.1 months versus 16.6 months. The control arm here is different than the control arm in the other trials, but that’s because there’s bevacizumab in PAOLA-1. [The PFS hazard ratio for all-comers was] 0.59 [95% CI, 0.49-0.72]. The BRCA-mutated group had a 37.2-month PFS [with olaparib] versus 17.7 months [without].
Can you go into more detail on SOLO-1?
SOLO-1 patients had a BRCA1 or BRCA2 mutation, [their disease was] newly diagnosed, they had to have [completed] a platinum-based therapy and...to have responded [to that therapy].3 They could have either neoadjuvant or primary debulking, so there were some patients who had no evidence of disease when they were randomized. It was 2:1 randomization, olaparib 300 mg twice a day versus placebo. The primary end point was investigator-assessed PFS. It was about a 400-patient study and...most patients had no evidence of disease, good performance status, and cancer antigen 125 within normal limits.
[Data on] PFS at 5-year follow-up were recently presented at the 2020 European Society for Medical Oncology meeting.6 We were awed by it. The median PFS had not been reached in the olaparib arm, and a lot of us were concerned that, with time, the curves would come together. These data, I think, we all found reassuring. We now have median PFS at 56 months versus 13.8 months [for olaparib vs placebo], 42 months’ difference between these; really spectacular.
The way the trial was designed, they had 2 years of maintenance therapy by olaparib. So at 5-year follow-up, the olaparib was stopped. There were a few patients that went on, but the vast majority of them stopped there, which is [why] a lot of us were afraid the curves would come together. But the difference between these curves was maintained. So whatever olaparib is doing, it looks like it has a continued impact; all of the subgroups benefited.3
The treatment-associated AEs [had] the class effects like nausea, fatigue, vomiting, and arthralgias. These were frequent but low grade. The nausea was almost all low grade, fatigue was fairly low grade, so these are manageable issues. There were some high-grade events of anemia, but again, usually manageable by transfusion or even dose reduction.
How do you decide when to use PARP inhibitors in your practice? How do mutations affect this decision?
The whole field has exploded with the fact that we think there’s a much larger group of patients who benefit from PARP inhibitors depending upon how you assess them. I’m a big sequencer. I still do a lot of sequencing, but I have certainly migrated to panel sequencing. But one can look at the patients who might benefit from PARP by looking at the effect of genomic instability, and that’s what Myriad myChoice CDx and Foundation Medicine are looking at.
Those 2 assays are almost all based on loss of heterozygosity [LOH], but the Myriad myChoice CDx adds the LOH assay telomeric allelic imbalance, which is measuring the imbalance on the chromosome. [This] is a little hard to measure by LOH, and it also adds large-scale state transitions. Sometimes the whole chromosome arm of the chromosome is shifted. If the arm of the chromosome is shifted, then LOH doesn’t tell you anything; it’s just in a different part of the genome. So looking at large scales adds something to this assay. The Foundation Medicine assay is a straight LOH assay.
[Genomic instability status (GIS)] measures the loss of DNA throughout the entire genome, the telomeric allelic imbalance, and large-scale state transitions. The Myriad assay adds up these scores and gives you the GIS—there’s a bit of a mystery how they add that, but nevertheless, you get this score.
The reason why it’s important is, if you look at BRCA mutations, there is a total contribution to HRD in ovarian cancer by gene mutation. BRCA1 and BRCA2 are the biggest contributors. But all the genes in the Fanconi DNA repair pathway contribute to DNA repair, and if they’re mutated, they’ll contribute to HRD. If you look at HRD assays, BRCA1 and BRCA2 account for about 20%, other homologous repair genes make up another 7%, and then there’s HRD from things like methylation or micro RNA. So we think 50% of patients with ovarian cancer have a defect in HRD. If you’re only sequencing...you may miss [some].
Thus, it’s not cut-and-dry, it’s a continuous variable. These assays have a cutoff, and so it’s not a perfect assay. You’re going to miss some patients that have genomic instability, and you’re going to throw in some patients who may not have a lot of genomic stability; so, not a perfect assay.
What do the National Comprehensive Cancer Network (NCCN) guidelines recommend here?
In the absence of BRCA1/2, the NCCN guidelines now say HRD status may provide information on the magnitude of benefit for PARP inhibitors.7 There is an issue of detecting genomic instability by HRD, and it may have an impact on whether the patient will benefit from PARP inhibitors.
What were the details of the PRIMA data?
The PRIMA study took all-comers and it was a double-blind, randomized, placebo-controlled phase 3 trial and was designed for niraparib [Zejula] versus placebo.4 Patients had to respond to platinum therapy. Response to platinum may be the best surrogate for PARP benefit.
I’ll point out a bit of a complication. Thrombocytopenia is a problem in certain patients with niraparib and in the middle of this study, they applied the “weights and plates,” which was developed based on the NOVA trial [NCT01847274].8 NOVA was the same study but for recurrent disease, where we found that those patients who had bad thrombocytopenia were patients with small body mass index and low platelet counts. So in the middle of this study, they applied weight and platelet counts, and those patients who were positive by this score were reduced to 200 mg [of niraparib] or started on 200 mg, and they eliminated a lot of thrombocytopenia.
The HRD and BRCA-mutated patient population had a hazard ratio of 0.40 [95% CI, 0.27-0.62]. You don’t need a statistician to tell you that niraparib is helping these patients. In the HRD-positive, BRCA wild-type population...the hazard ratio wasn’t quite as good, at 0.50 [95% CI, 0.31-0.83]. The homologous repair proficient [HRP] was probably the biggest [issue] about this study, which is that you would predict these patients shouldn’t benefit, but they do. It was statistically significant with a hazard ratio of 0.68 [95% CI, 0.49-0.94], but the difference is pretty small, about 3.5 months.
The overall primary end point of PFS in the overall population [had a hazard ratio of] 0.62 [95% CI, 0.50-0.76; P <.001] in favor of niraparib, 13.8-month median PFS versus 8.2 months.
The treatment-associated AEs are a common theme, which are nausea, fatigue, some vomiting—all low grade. The difference is this thrombocytopenia; PRIMA didn’t make it easy for us to interpret this. They had thrombocytopenia, but they also had decreased platelet counts because the investigators separated them based on the case report forms. But the bottom line is, even though they used weight and platelet counts, there was still thrombocytopenia associated with niraparib, which is why the FDA said [on the label that] the first month you need to get platelet counts every week.
That led...to the FDA approval for niraparib in first-line maintenance of patients with advanced ovarian cancer.9 The indication was for all-comers, and that was in the face of having olaparib approved for SOLO-1 only for the BRCA-mutated population.10
Please describe the role PAOLA-1 played in this setting.
PAOLA-1, I think, has given us essentially another choice.5 It is primarily a European study that [looked at] frontline treatment. Patients had to respond to platinum therapy with either complete or partial response, but they could have no evidence of disease based on surgery. They then were randomized to olaparib plus bevacizumab versus bevacizumab alone. The reason this was done in Europe is because based upon GOG-0218, many patients in Europe get bevacizumab. So they decided to compare bevacizumab versus the combination bevacizumab plus olaparib, with the primary end point of investigator-initiated PFS.
The overall PFS with olaparib plus bevacizumab was clearly superior to bevacizumab alone, with a hazard ratio of 0.59 [95% CI, 0.49-0.72; P < .001], which is not as good as PRIMA or SOLO-1. But remember, your control arm is being treated, and that probably has something to do with it. This was highly statistically significant.
If you look at the tumor BRCA-mutation status, it looks awfully familiar to that of SOLO-1, with a hazard ratio of 0.31 [95% CI, 0.20-0.47]. The combination works very well in patients with BRCA mutations. If you look at non–BRCA-mutated tumors—but these are going to have some HRD patients in it—there was still a separation of curve. The hazard ratio was not quite as good, at 0.71 [95% CI, 0.58-0.88].
Probably the biggest difference about PAOLA-1...was for patients with HRD, including tumor-mutated BRCA. There was a big separation [of curves]. We’d expect this hazard ratio of 0.33 [95% CI, 0.25-0.45]. For [patients who were] HRD positive without the BRCA mutation, there was still a big difference. The hazard ratio was not as good, but if you look at the HRP and unknown population, you see no effect with a hazard ratio of 0.92; it overlaps to 1.00 [95% CI, 0.72-1.17].
The safety analysis was similar to what I’ve said before except there was hypertension. You would expect [this] because you have bevacizumab in the regimen and that’s going to give patients hypertension. The amount of acute myeloid leukemia or myelodysplastic syndrome was not significant; neither were new primaries or even pneumonitis. So that was reassuring.
PRIMA was approved at roughly the same time the FDA approved the use of olaparib and bevacizumab in these tumors. But I will point out, the label is only for HRD tumors and not HRP, and that was because of the PAOLA-1 data.11
How does the tolerability compare between SOLO-1, PAOLA-1, and PRIMA?
There were a lot of AEs in these trials, but most were low grade. If you look at the high-grade AEs, they were roughly the same with the exception of PRIMA, and that was probably because of the platelet issue.
More importantly than AEs though, I think, is looking at how often doses are interrupted, how often doses are reduced, and how often [patients] discontinued. Dose interruptions were not terribly different, with the exception of niraparib—again, this is the platelet issue. Dose reductions were reasonably consistent, but fairly high in niraparib because of this issue of some patients having thrombocytopenia, which I think has been somewhat solved by weight and platelet counts. Discontinuations were interesting, similar in SOLO-1 to PRIMA. So, even though there was a reduction, there wasn’t [a lot of] discontinuation. It was a little higher with the combination of bevacizumab and olaparib, and it’s not clear to me which drug is contributing to it.
What is the best way to modify the therapeutic doses?
Niraparib has a bit of an advantage because [it comes in] 100-mg capsules; you’re using 2 or 3 of them, and in order to dose reduce, you just drop a capsule. Also, because it’s a 1-a-day drug, if your patient has nausea, they can take it at night so they sleep through it. Olaparib and rucaparib are twice-daily drugs. I’ve had patients on rucaparib whom I’ve dose reduced and [for whom I] then had to write a new script, which is a pain, and I’ve had a couple of patients get charged 2 co-pays.
What would you pick from this poll?

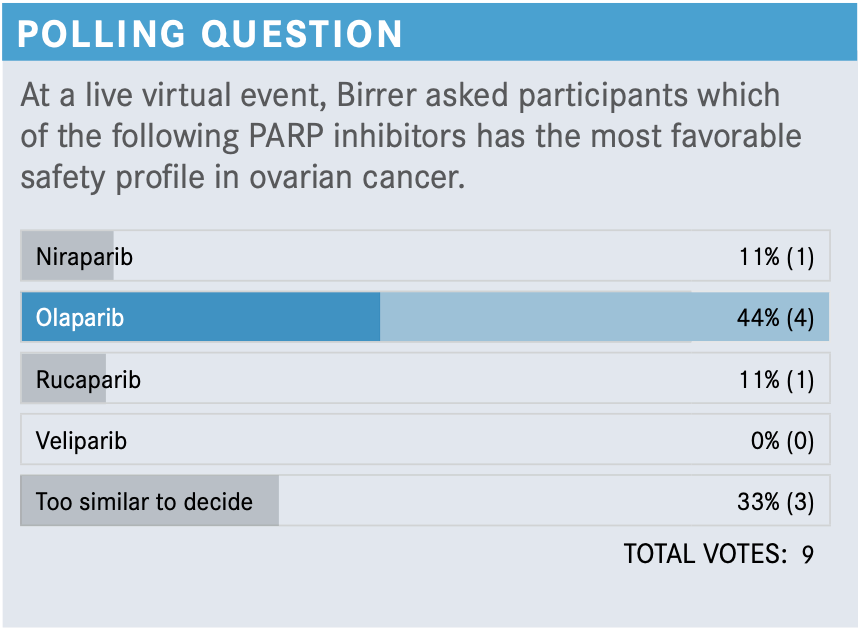
I think that they’re all very safe even in these subtle differences. For instance, niraparib with thrombocytopenia is manageable, but it is probably a slightly more complicated PARP inhibitor. As long as you recognize some of these issues, then you’ll be fine. If you were to ask me what the biggest problem is with giving PARP inhibitors, I would say it’s the fatigue. If you have patients who are on maintenance, who like to be active, and they have even grade 1 fatigue, that could be really [bothersome]. I've had some issues with that. I treat most of my patients who have that problem with methylphenidate [Ritalin] and about 50% of the time it works well; the other 50% of the time it works horribly. They feel very dysphoric, and so in those cases I’ll dose reduce.
In light of these data, where does the role of intraperitoneal (IP) chemotherapy fall at this point? Are you a fan or not, or what are you doing now?

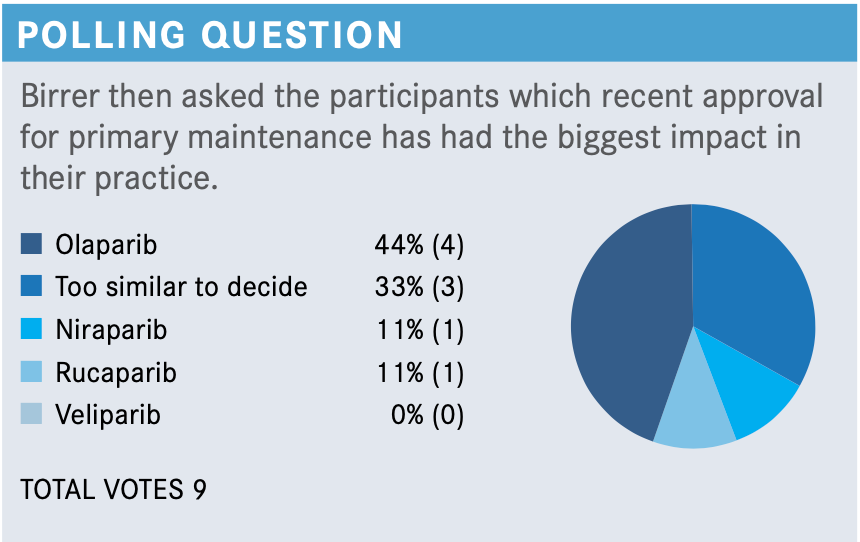
I’ve never been an IP fan, but I’ve worked with a lot of surgeons who have given it. I think with GOG-0252 [NCT00951496] coming out as negative and now having more options, including bevacizumab for up-front treatment, we’re seeing IP slowly die, but there are certain centers that are still doing it. I would not recommend IP to any of my patients for places that don’t use it a lot. You have to use it a lot to do it right.
REFERENCES
1.Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473-2483. doi:10.1056/NEJMoa1104390
2. Tewari KS, Burger RA, Enserro D, et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J Clin Oncol. 2019;37(26):2317- 2328. doi:10.1200/JCO.19.01009
3. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495-2505. doi:10.1056/NEJMoa1810858
4. González-Martín A, Pothuri B, Vergote I, et al; PRIMA/ENGOT-OV26/GOG-3012 Investigators. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391-2402. doi:10.1056/NEJMoa1910962
5. Ray-Coquard I, Pautier P, Pignata S, et al; PAOLA-1 Investigators. Olaparib plus beva- cizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416- 2428. doi:10.1056/NEJMoa1911361
6. Banerjee S, Moore KN, Colombo N, et al. Maintenance olaparib for patients (pts) with newly diagnosed, advanced ovarian cancer (OC) and a BRCA mutation (BRCAm): 5-year (y) follow-up (f/u) from SOLO1. Ann Oncol. 2020;31(suppl 4):S613. doi:10.1016/ annonc/annonc276
7. NCCN. Clinical Practice Guidelines in Oncology. Ovarian cancer: including fallopian tube cancer and primary peritoneal cancer, version 2.2020. Accessed January 20, 2021. https://bit.ly/2LItdBH
8. Berek JS, Matulonis UA, Peen U, et al. Safety and dose modification for patients receiving niraparib. Ann Oncol. 2018;29(8):1784-1792. doi:10.1093/annonc/mdy181
9. FDA approves niraparib for first-line maintenance of advanced ovarian cancer. FDA. April 29, 2020. Accessed January 20, 2021. https://bit.ly/35ZuM5Y
10. FDA approved olaparib (Lynparza, AstraZeneca Pharmaceuticals LP) for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated (gBRCAm or sBRCAm) advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based. FDA. Updated December 26, 2018. Accessed January 20, 2021. https://bit.ly/36iuNBl
11. FDA approves olaparib plus bevacizumab as maintenance treatment for ovarian, fallopian tube, or primary peritoneal cancers. FDA. Updated May 11, 2020. Accessed January 20, 2021. https://bit.ly/394VeMz











Survivorship Care Promotes Evidence-Based Approaches for Quality of Life and Beyond
March 21st 2025Frank J. Penedo, PhD, explains the challenges of survivorship care for patients with cancer and how he implements programs to support patients’ emotional, physical, and practical needs.
Read More

