SHP2 Inhibitors Undergo Exploration in Combinations
Melissa Johnson, MD, discussed SHP2 inhibitors with Targeted Therapies in Oncology.



SHP2 inhibition remains a focus of ongoing research because of its role in signaling pathways across cancer settings, but an SHP2 inhibitor remains elusive. Melissa Johnson, MD, director of lung cancer research at Sarah Cannon Research Institute in Nashville, Tennessee, discussed SHP2 inhibitors with Targeted Therapies in Oncology.
Johnson said that SHP2 inhibitor therapy “is an area of active investigation—none of the compounds being evaluated have been approved by the FDA. There are a couple of critical questions that we are asking about this emerging class of agents. The first question is, ‘Are SHP2 inhibitors something that we can add when patients develop resistance to a particular therapy?’ And, second, ‘Should these therapies be added from the beginning to prolong the time until resistance develops in the first place?’”
Proto-oncogene PTPN11 encodes SHP2, which, as a protein tyrosine phosphatase, is involved in posttranslational modification and plays a role in myriad cell functions. SHP2 is positioned at a convergence of several signaling pathways. Consequently, SHP2 serves varying functions with substrate specificity depending upon the signaling cascade. Such pathways involve cell survival and immune regulation, and evidence has shown that SHP2 dysregulation is associated with hematologic and solid tumor malignancies. SHP2 can activate the RAS-RAF-MEK-ERK signaling pathway and both stimulate or antagonize the PI3K-AKT and JAK-STAT pathways. In addition, SHP2 inhibits T cells via the PD-1/PD-L1 pathway. Because of the various SHP2 functions, both activating and inhibiting agents have been developed to target SHP2.1,2
In clinical oncology, multiple SHP2 inhibitors are in development.1,2 SHP2 is downstream of a receptor tyrosine kinase (RTK), which activates several signaling pathways through which cancer cells may become drug resistant. For example, genetic mutation affecting EGFR, a variety of RTK, activates pathways leading to drug resistance induction. Also, SHP2 upregulation has been reported during RTK activation as part of acquired drug resistance. To that end, hepatoma cells treated with a multikinase inhibitor also targeting RTK show reactivation of RAS-RAF-MEK-ERK and AKT signaling pathways. But when combined with a SHP2 inhibitor, the MEK/ERK and AKT pathway reactivation is blocked, thereby bypassing the kinase inhibitor resistance.1

SHP2 Inhibitors as Monotherapy
In Hematologic Malignancies
Acquired resistance to tyrosine kinase inhibitor (TKI) therapy remains a challenge in the treatment of patients with hematologic malignancies. Chronic myeloid leukemia (CML) and Philadelphia chromosome-positive (Ph+) B-cell lymphoblastic leukemia (B-ALL) are caused by isoforms of constitutively activated tyrosine kinase BCR-ABL1, resulting from the BCR-ABL1 oncogene. Although TKIs targeted to BCR-ABL1 have improved the treatment landscape of these hematologic malignancies, many patients with CML who receive this treatment remain at risk of relapse because of CML stem cell insensitivity to the TKIs.3 The response to TKIs is worse still in patients with Ph+ B-ALL, and multiple mechanisms of TKI resistance have been suggested. One study model recently showed that SHP2 is necessary for BCR-ABL1–initiated lymphoid and myeloid neoplasia.3 Additional studies have shown that SHP2 is involved in mul- tiple pathways associated with hematologic malignancy oncogenesis. Although several SHP2 inhibitors are undergoing investigation in clinical trials, they will require additional development to counteract resistance to drugs used in the treatment of patients with hematologic malignancies.4
In Solid Tumors
A pattern similar to that of SHP2 expression in hematologic malignancies has been reported in tumor cells. For example, SHP2 pathways are activated and promote cancer progression in breast cancer cells. Also, in patients with pancreatic ductal adenocarcinoma, higher levels of SHP2 expression are associated with poorer outcomes than are lower levels of SHP2 expression.5 Furthermore, in a KRAS-mutant non–small cell lung cancer (NSCLC) xenograft model, SHP2 inhibition appeared to induce tumor cell senescence and impair tumor growth.6 Similarly, in breast cancer cells, SHP2 inhibition was observed to suppress tumorigenesis and induce a near-normal cellular phenotype.7 Ongoing clinical trials include a phase 1 trial of SHP2 inhibitor TNO155 alone or combined with EGFR TKI nazartinib in patients with advanced solid tumors (NCT03114319) and a phase 1 trial of SHP2 inhibitor ET0038 as monotherapy in patients with advanced solid tumors (NCT05354843).
SHP2 Inhibitors in Combination Therapy
“We are evaluating SHP2 inhibitors with a number of other targeted therapies such as KRAS-G12C inhibitors, EGFR inhibitors, BRAF inhibitors, MEK inhibitors, and even PD-L1 inhibitors,” Johnson said. “In general, we know that when SHP2 is dephosphorylated, the MAPK pathway is activated. Thus, when SHP2 activity is inhibited, signaling via RTKs such as EGFR may be hindered, as well as downstream effectors such as KRAS and ERK.”
With MEK Inhibitors
Although RAS-mutated tumors are often treated with MEK inhibitors, MEK mutation results in excessively activated MEK or prevents inhibitor binding, thereby causing MEK inhibitor resistance. However, research has shown that SHP2 inhibitors can prevent adaptive MEK inhibitor resistance. Hence, combining MEK and SHP2 inhibitors is a proposed strategy for RAS-mutant malignancies. In various tumor cell lines, combined MEK and SHP2 inhibitors have been shown to inhibit or prevent tumor growth, promote tumor regression, and show greater sensitivity to the MEK inhibitor.1,8,9
With ERK Signal Suppressors
In ERK-dependent tumors, the disruption of RAF or MEK negative feedback leads to RTK upregulation, resulting in RAS activation and a return of ERK activity, and with that, inhibitor resistance. Combining ERK, MEK, and SHP2 inhibitors has been shown to suppress ERK and SHP2 signals in RKO xenografts.1 For example, use of ERK inhibition with dabrafenib (Tafinlar) and trametinib (Mekinist) and SHP2 inhibition with SHP099 in an RKO xenograft model suppressed tumor growth. Such signal suppression may also manage BRAF V600E–mutated colorectal tumors.1,10
With ALK Inhibitors
As with tumors that develop MEK inhibitor-resistance, NSCLC with ALK rearrangements may develop resistance to ALK inhibitors. In ALK inhibitor–resistant cells that underwent short hairpin RNA gene screening, SHP2 was identified as a resistance node. When SHP2 inhibitor SHP099 was combined with ALK TKI ceritinib (Zykadia), the growth of resistant patient- derived tumor cells was inhibited.11
With PD-1/PD-L1 Targeted Agents
“Another area of investigation is combining SHP2 inhibitors with PD-1 or PD-L1 inhibitors, and the science behind this combination is also interesting to me,” Johnson said. “SHP2 seems to also be a mediator of PD-L1 inhibition of T-cell function by inactivating its coreceptor CD28. Therefore, using these drugs in combination may be a more orthogonal approach to reversing immunosuppression within the tumor microenvironment that has been evaluated previously.”
In addition to being downstream of an RTK, SHP2 is also downstream in the PD-1 pathway, which is involved in T-cell suppression and anergy.12 Moreover, SHP2 can inhibit immune cell activation through binding various immunosuppressive receptors. Hence, SHP2 inhibitors have shown potent tumoricidal activity.1 When SHP2 inhibitor SHP099 was combined with a PD-1 inhibitor in xenograft models of colon cancer, tumor growth was better controlled than it was in response to each inhibitor as a monotherapy.12
But the use of a SHP2 inhibitor as part of a combination regimen is challenging in terms of both efficacy and safety, according to Johnson. “The truth is, SHP2 inhibitors have to be given with another therapy in order to be effective,” Johnson said. However, she explained, “we haven’t been able to do that very successfully yet because [SHP2 inhibitors are] not well tolerated on their own. SHP2 inhibitors have been evaluated in a variety of schedules to improve this tolerability—2 days on, several days off, or 1 big dose once per week; or 3 days on, 4 days off, or even 2 weeks on, 1 week off.”
Because SHP2 inhibitors would potentially add toxicity to “RTK therapies that, for the most part, have satisfactory tolerability profiles,” such as an EGFR inhibitor, Johnson explained, “the combination strategy results in more [adverse] effects. That trade-off has to be worth it. It [had] better prolong the amount of time that patients can be on both therapies in combination. I do not know that we have figured out how to do that yet.”
The Future of SHP2 Inhibitors
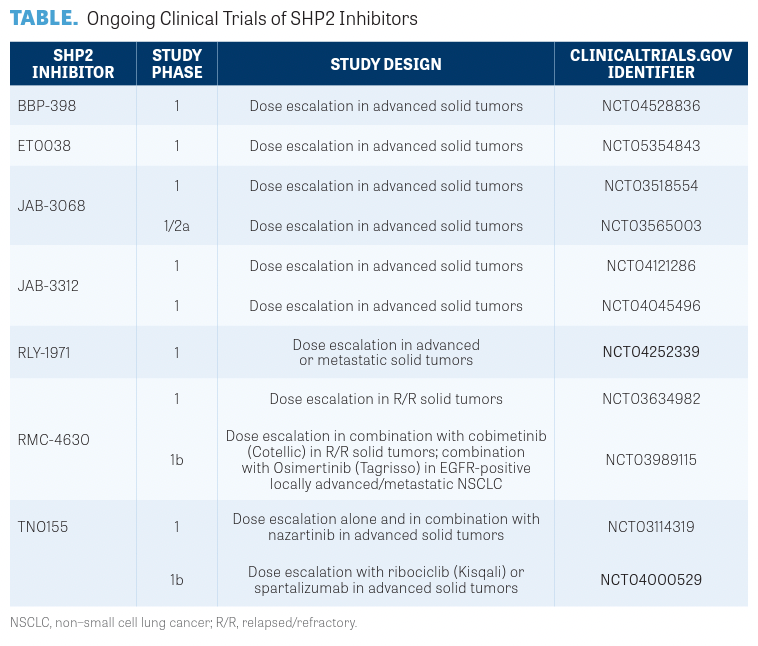
“The other mechanism that makes a lot of sense is in KRAS G12C inhibitors,” Johnson said. “When the KRAS G12C inhibitors bind with the KRAS gene, they are locked in the off position and can no longer signal mutated cancer cells to grow. Therefore, adding a SHP2 inhibitor would seem to shift more of the KRAS gene to its GDP-bound off state where the KRAS off inhibitor can bind.”13 For a summary of the various SHP2 inhibitors in development, please refer to the TABLE.
Overall, the safety profiles of the SHP2 inhibitors undergoing development are comparable, according to Johnson. “In my opinion, [the SHP2 inhibitors] have very similar mechanisms of action, with similar [adverse] effect profiles that include peripheral and pulmonary edema, decreases in ejection fraction, and some cytopenias,” Johnson said.
Although it’s difficult to forecast when SHP2 inhibitors may enter the physician’s armamentarium, SHP2 inhibition could address several unmet needs, according to Johnson. “The goal of a SHP2 inhibitor [would be] either that it prolongs the amount of time until treatment resistance develops in the first place, or it reactivates a pathway that has become resistant with a monotherapy- targeted approach,” Johnson said. “Some other unmet needs, though, are that these patients with oncogene-driven cancers are the ones who develop brain metastases, so if these SHP2 inhibitors were particularly active in the CNS [central nervous system], that would be another way to win. However, some have been designed particularly with CNS penetration in mind.” Ultimately, Johnson said, “I think that the SHP2 inhibitor with the best tolerability profile and therefore ease of combining with another targeted therapy will be the winner.”
REFERENCES
1. Liu M, Gao S, Elhassan RM, Hou X, Fang H. Strategies to overcome drug resistance using SHP2 inhibitors. Acta Pharm Sin B. 2021;11(12):3908-3924. doi:10.1016/j. apsb.2021.03.037
2. Song Y, Zhao M, Zhang H, Yu B. Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol Ther. 2022;230:107966. doi:10.1016/j.pharmthera.2021.107966
3. Gu S, Sayad A, Chan G, et al. SHP2 is required for BCR-ABL1-induced hematologic neoplasia. Leukemia. 2018;32(1):203-213. doi:10.1038/leu.2017.250
4. Kanumuri R, Kumar Pasupuleti SK, Burns SS, Ramdas B, Kapur R. Targeting SHP2 phosphatase in hematological malignancies. Expert Opin Ther Targets. 2022;26(4):319-332. doi:10.1080/14728222.2022.2066518
5. Zheng J, Huang S, Huang Y, et al. Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumour Biol. 2016;37(6):7853-7859. doi:10.1007/ s13277-015-4675-5
6. Ruess DA, Heynen GJ, Ciecielski KJ, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954-960. doi:10.1038/s41591-0180024-8
7. Song Z, Wang M, Ge Y, et al. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm Sin B. 2021;11(1):13-29. doi:10.1016/j.apsb.2020.07.010
8. Mainardi S, Mulero-Sánchez A, Prahallad A, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24(7):961-967. doi:10.1038/ s41591-018-0023-9
9. Torres-Ayuso P, Brognard J. Shipping out MEK inhibitor resistance with SHP2 inhibitors. Cancer Discov. 2018;8(10):1210-1212. doi:10.1158/2159-8290.CD-18-0915
10. Ahmed TA, Adamopoulos C, Karoulia Z, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019;26(1):65-78.e5. doi:10.1016/j.celrep.2018.12.013
11. Dardaei L, Wang HQ, Singh M, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. 2018;24(4):512517. doi:10.1038/nm.4497
12. Zhao M, Guo W, Wu Y, et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm Sin B. 2019;9(2):304-315. doi:10.1016/j. apsb.2018.08.009
13. Mukhopadhyay S, Fedele C, Neel BG. SHP2 inhibitors for treating cancer. National Cancer Institute. April 15, 2021. Accessed April 26, 2023. https://bit.ly/3oMdggN

















