Treatment Strategies to Optimize Outcomes With Brentuximab Vedotin: Management of Common and Rare Toxicities
Common AEs associated with brentuximab vedotin, such as peripheral neuropathy and neutropenia, are often manageable with modifications to dose and schedule.


Michelle A. Fanale, MD

Michelle A. Fanale, MD
University of Texas
MD Anderson Cancer Center
Houston

Nancy C. Whiting, PharmD

Nancy C. Whiting, PharmD
Seattle Genetics, Inc
Bothell, Washington

Ellen Neylon, RN

Ellen Neylon, RN
Columbia University Center
for Lymphoid Malignancies
New York, New York
Abstract
Background: Brentuximab vedotin is an antibody-drug conjugate that targets CD30-expressing cells and has demonstrated single-agent clinical efficacy in pivotal phase II trials in relapsed or refractory Hodgkin lymphoma after autologous stem cell transplant and in relapsed or refractory systemic anaplastic large cell lymphoma. Brentuximab vedotin carries an adverse event (AE) profile that differs from those of traditional monoclonal antibody therapies. Herein we discuss the management of common AEs in addition to symptoms that may accompany rare AEs associated with brentuximab vedotin.
Materials and Methods:Published, sponsored clinical trial studies of single-agent brentuximab vedotin were surveyed to identify the most commonly reported AEs. Reporting rates for rare AEs were estimated from cases drawn from the Seattle Genetics drug safety database using the current version of the Medical Dictionary for Regulatory Activities.
Results: Peripheral neuropathy and neutropenia were identified as the most frequently reported AEs across the published clinical trials. Rare AEs included progressive multifocal leukoencephalopathy, Stevens-Johnson syndrome/toxic epidermal necrolysis, pancreatitis, pulmonary toxicity, and hepatotoxicity; reporting rates for these rare AEs (<2%) were within or below population-specific background ranges reported in the literature.
Conclusion: Common AEs associated with brentuximab vedotin, such as peripheral neuropathy and neutropenia, are often manageable with modifications to dose and schedule. While rare AEs have been reported at rates similar to or below background, vigilant monitoring is necessary to detect and address these serious potential complications.
Introduction
The antibody-drug conjugate (ADC) brentuximab vedotin is composed of a CD30-specific monoclonal antibody, cAC10, attached to the microtubule disrupting agent, monomethyl auristatin E (MMAE), via a protease-cleavable dipeptide linker.1,2Brentuximab vedotin binds to and is internalized by CD30-expressing cells, where it is trafficked to the lysosome. The dipeptide linker is selectively cleaved within the lysosome, releasing MMAE into the cell, where it binds microtubules and disrupts the microtubule network, inducing G2/M cell cycle arrest and apoptosis.1,3,4
Single-agent brentuximab vedotin was studied at a dose of 1.8 mg/kg administered as a 30-minute intravenous infusion every 3 weeks in 2 multinational, single-arm, open-label, phase II pivotal trials.5,6In one trial, brentuximab vedotin was administered to 102 patients with relapsed or refractory Hodgkin lymphoma (HL) after autologous stem cell transplant. In this study, 75% of patients achieved an objective response (n = 76; 95% confidence interval [CI], 64.9-82.6), with a complete remission (CR) rate of 34% (n = 35; 95% CI, 25.2-44.4). Another pivotal phase II trial was conducted to evaluate single-agent brentuximab vedotin in 58 patients with relapsed or refractory systemic anaplastic large cell lymphoma (sALCL). In that study, 86% of patients achieved an objective response (95% CI, 74.6-93.9), with a CR rate of 57% (95% CI, 43.2- 69.8). Efficacy results for both trials are summarized inTABLE 1.
Table 1. Brentuximab Vedotin Clinical Trials: Design, Baseline Patient Characteristics, and Efficacy Outcomes [CLICK TO DOWNLOAD]
Because it contains a cytotoxic agent, the adverse events (AEs) profile of brentuximab vedotin differs from that of unconjugated monoclonal antibodies. Appropriate surveillance and management of the AEs associated with brentuximab vedotin are important to enable patients to benefit fully from this novel therapy. The objective of this publication is to outline strategies for the management of common and rare AEs that may arise during treatment with brentuximab vedotin.
Case Study
We present a 67-year-old female with CD30-positive diffuse large B-cell lymphoma, previously treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) and RICE (rituximab, ifosfamide, carboplatin, etoposide), who could not undergo autologous stem cell transplant (ASCT) because of insufficient stem cell collection. The patient’s disease relapsed 2 months after chemotherapy, and she enrolled in a clinical study of brentuximab vedotin. Additional complicating medical issues included polymyositis on active steroid therapy, documented stage III nonalcohol steatohepatitis, and hypogammaglobulinemia on replacement intravenous immunoglobulin (IVIG) treatment. The patient received 2 cycles of brentuximab vedotin at a dose of 1.8 mg/kg and achieved a CR after the second cycle. She also experienced infusion-related reactions (IRRs), including shortness of breath, and was subsequently premedicated with hydrocortisone, diphenhydramine, famotidine, and acetaminophen.
The patient was unable to continue treatment on the clinical study because of persistently elevated transaminases, and she experienced disease progression 3 months after her removal from the study. Upon enrollment in a retreatment study, the patient was treated with 1.8 mg/kg brentuximab vedotin and again achieved CR after the second cycle. The dose was reduced to 1.2 mg/kg after the third cycle because of repeat elevation of transaminases. After dose reduction, peak transaminases were reduced from approximately 400 U/L to the mid-100 U/L range. The patient entered observation after completing 11 cycles on this protocol.
The patient’s disease progressed 3 months after she stopped therapy. She is currently being treated with brentuximab vedotin, off protocol, at a dose of 1.2 mg/kg. The patient again experienced elevated transaminases, along with worsening of peripheral sensory neuropathy from grade 1 to grade 2/3. By lengthening the dosing interval from 3 weeks to 5 weeks, and subsequently further, transaminase levels improved to the 100 U/L range, and peripheral neuropathy improved to grade 1. To date, the patient has completed 20 cycles off protocol. At the time of this publication, the patient was being treated with brentuximab vedotin for an off-label indication in the United States.
Materials And Methods
We surveyed published, sponsored clinical trial data to identify the most frequently reported AEs associated with single-agent use of brentuximab vedotin. These studies included the phase II pivotal trials in relapsed or refractory HL and sALCL, a phase II study of extended treatment in relapsed or refractory CD30-positive hematologic malignancies, and a phase II study in patients with T-cell lymphoma (TABLE 1). Common AEs that occurred with a frequency of at least 25% in any study were included in this report. To estimate reporting rates for serious AEs, we tabulated cases drawn from the Seattle Genetics drug safety database (DSB) using the current version of the Medical Dictionary for Regulatory Activities (MedDRA) at the time of each search. Search strategies included Standardized MedDRA Queries (SMQs), where available, as well as custom search queries. Search results include all safety reports in the DSB up to August 18, 2014.
Recommendations for surveillance and management of AEs were drawn from internal databases and from real-life practices based on our clinical experiences with brentuximab vedotin. Unless otherwise noted, recommendations on dose modifications reflect the current US Prescribing Information (USPI); guidance may vary in other regions.
Results
Peripheral neuropathy
Common adverse events associated withbrentuximab vedotin
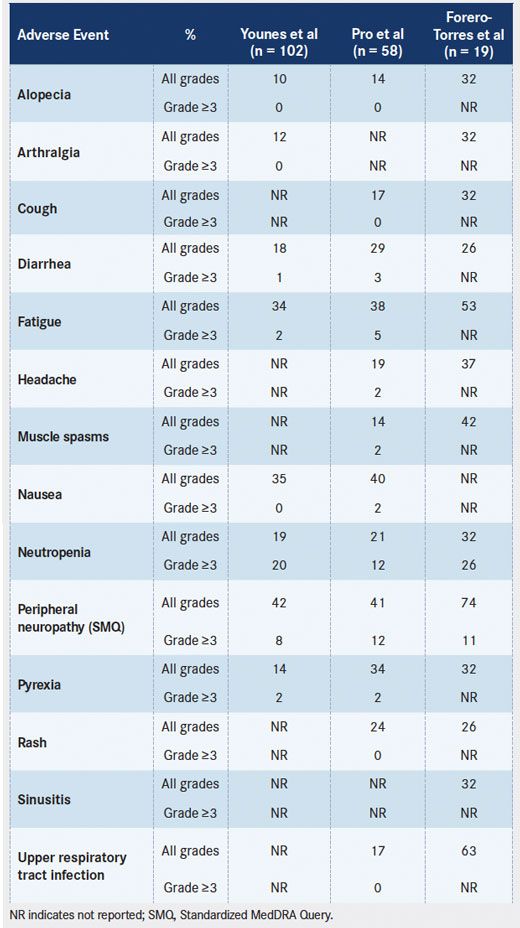
Peripheral neuropathy was the most commonly reported AE in most published clinical trials on brentuximab vedotin (TABLE 2). Peripheral neuropathy, as identified by SMQ, was reported in approximately 55% of patients and was the primary cause of treatment-related discontinuations in the pivotal phase II trials in HL and sALCL.5,6Overall, 10% to 15% of patients experienced grade 3 events; no grade 4 events were reported. The peripheral neuropathy reported in the pivotal trials was cumulative and primarily sensory in nature, although motor neuropathy was experienced by 13% of patients (Seattle Genetics, Inc, unpublished data). Preexisting peripheral neuropathy, primarily grade 1, was present in 24% of patients in these studies. In the pivotal trials, the median time to first onset of any peripheral neuropathy was 12.4 weeks (approximately 4 cycles). Peripheral neuropathy (SMQ) led to dose delays in 15% of patients, to dose reductions in 9% of patients, and to treatment discontinuations in 12% of patients across the 2 trials. Partial improvement (by 1 grade or more) or complete resolution of peripheral neuropathy events after dose holding or dose reduction were reported in approximately 80% of patients, with a median time to improvement or resolution of 13.2 weeks and 9.9 weeks in HL and sALCL, respectively.5,6 Peripheral neuropathy was also observed in other published clinical trials (TABLE 2).
TABLE 2. Drug-Related Adverse Events of Brentuximab Vedotin Occurring in ≥25% of Patients in Any Published Clinical Trial


Drug-Related Adverse Events of Brentuximab Vedotin Occurring in more than 25 percent of Patients in Any Published Clinical Trial
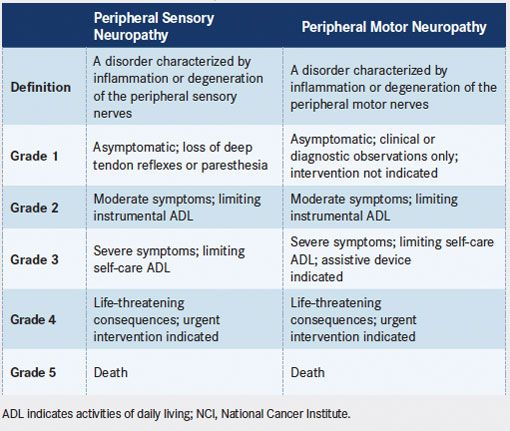
Risk factors for peripheral neuropathy include diabetes mellitus, vitamin B deficiency, glutamine deficiency, hypothyroidism (commonly seen in patients after field irradiation), and prior or concomitant use of other treatments associated with peripheral neuropathy.7-10Patients who experience symptoms of peripheral neuropathy may describe tingling or a feeling of numbness that starts at the tips of their fingers or toes. Some patients may also report muscle aches or overall muscle weakness that is related to neuropathy. Appropriate assessment and documentation of preexisting peripheral neuropathy before the start of therapy are essential to establish baseline neurologic status. Current grading of peripheral neuropathy, as defined in the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, is listed inTABLE 3. Note that these criteria differ from those listed in version 3.0, which was used to assess neuropathy in the pivotal trials. Neuropathy should be assessed before administration of every dose, and assessments should remain consistent among examinations. However, it is often useful to introduce new tasks during assessments so that deficits may be identified. For example, a patient who is able to fasten buttons at each examination may be unable to pick up a coin from a flat surface or to write legibly. Patients should be educated on symptoms of peripheral neuropathy, and it may be useful to remind them of symptoms reported at baseline for comparison to their current status.
TABLE 3. Definitions and Grading of Peripheral Sensory and Motor Neuropathy (reproduced from NCI Common Terminology Criteria for Adverse Events, Version 4.03)

")
Definitions and Grading of Peripheral Sensory and Motor Neuropathy (reproduced from NCI Common Terminology Criteria for Adverse Events, Version 4.03)
The peripheral neuropathy associated with brentuximab vedotin is generally reversible and may often be managed with modifications to dosing and schedule. In patients who present with new or worsening grade 2 or 3 neuropathy, dosing should be held until neuropathy improves to grade 1 or to baseline and then restarted at 1.2 mg/kg. Patients who successfully resume treatment at the reduced dose should not undergo dose escalation to 1.8 mg/kg, because symptoms may return. Treatment should be discontinued in patients with grade 4 peripheral neuropathy. We also recommend that patient education on management of neuropathy include information on hydration and maintaining adequate electrolyte levels per institutional guidelines, in addition to guidance on keeping extremities warm.
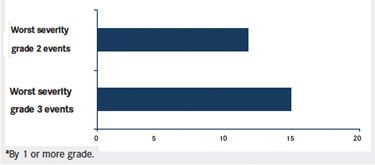
Both clinical trial and practical experience suggest that it is preferable to address peripheral neuropathy at lower grades of severity. Indeed, our experience is that neuropathy improves more quickly when dosing is held at the emergence of grade 2 neuropathy, before it worsens to grade 3. Dosing may then be resumed sooner, although at the reduced dose, which may result in better therapeutic effects. Data from the pivotal trials also support initiation of dose holding at the emergence of grade 2 rather than grade 3 neuropathy. In those trials, the median time to resolution or improvement of peripheral neuropathy events with a worst severity of grade 2 was 12.1 weeks compared with 15.6 weeks for events with a worst severity of grade 3 (FIGURE).
FIGURE. Time to resolution or improvementaof peripheral neuropathy events in phase II pivotal trials.


FIGURE. Time to resolution or improvement of peripheral neuropathy events in phase II pivotal trials
With patient education and proper management, peripheral neuropathy should not prevent patients from receiving treatment to disease progression. In an extension study of patients with relapsed or refractory CD30-positive hematologic malignancies, 19 patients received a median of 24 cycles of brentuximab vedotin (range, 17-42) over a median of 21 months (range, 14-32).11Peripheral sensory neuropathy was reported in 74% of patients, with first onset occurring within cycles 1 through 16 in 53% of patients. Dose reductions and delays per protocol occurred in 37% and 74% of patients, respectively.
Based on our clinical experience, alterations in dosing interval may be considered after resumption of dosing at 1.2 mg/kg, particularly for patients in CR. Lengthening the dosing interval by 1 or more weeks, for example, may minimize the risk of recurrence of grade 2 or higher peripheral neuropathy events while allowing the patient to remain on treatment. It is important to note, however, that this recommendation is not detailed in the prescribing information.
Neutropenia
Neutropenia was commonly reported across the published clinical trials and was a primary cause of dose delays (TABLE 2). In the pivotal phase II trials in HL and sALCL, neutropenia, predominantly grade 3 or higher, was reported in 19% of patients and led to dose delays in 15% of patients.5,6The neutropenia observed in the trials was transient and did not contribute to cases of febrile neutropenia, although febrile neutropenia has been reported in postmarketing experience.
Surveillance is again crucial to ensuring safe and appropriate treatment and should include monitoring of complete blood counts prior to each dose of brentuximab vedotin. In patients with grade 3 or 4 neutropenia, more frequent monitoring is recommended. If grade 3 or 4 neutropenia develops, dosing should be suspended until neutropenia resolves to baseline or less than or equal to grade 2, and subsequently resumed at a dose of 1.2 mg/kg. In practice, we find that neutropenia often resolves by the time of the patient’s next dose on the standard 21-day schedule; delays are typically brief. However, for the first few doses it may be useful to monitor complete blood counts mid-cycle to guard against prolonged instances of early neutropenia. Growth factor support for subsequent doses may be considered but, in our experience, has not frequently been needed.
Rash
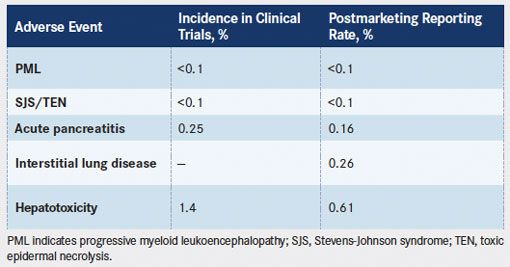
Rash was a common AE reported in the phase II and other clinical trials. Rash, grades 1 and 2, was reported in 27% of patients in the phase II HL trial and in 31% of patients in the phase II sALCL trial.12Stevens-Johnson syndrome (SJS) was reported in 1 patient in the pivotal trials and, in combination with toxic epidermal necrolysis (TEN), occurs at a rate of less than 0.1%, accounting for all AE reporting sources (TABLE 4). Nearly all of these cases were confounded by concomitant drugs known to cause SJS/TEN, including co-trimoxazole and allopurinol, as well as underlying clinical conditions such as systemic cancer and ongoing infections (Seattle Genetics, Inc, unpublished data). Rash was reported in 10% of patients in an ongoing, open-label, phase II trial evaluating brentuximab vedotin in patients with CD30-positive non-Hodgkin lymphoma.13Grade 3 macular rash was also reported in 1 patient on the same trial.14Rash was reported in 27% of patients in an open-label, phase II trial evaluating brentuximab vedotin in CD30-positive lymphoproliferative disorders and in CD30-positive mycosis fungoides.15
TABLE 4. Rare Adverse Event Rates


Rare Adverse Event Rates
In practice, patients may present with mild rash that is observed more frequently in sALCL than in HL. Symptomatic management with topical 1% hydrocortisone or a short course of systemic corticosteroids (eg, methylprednisolone) is often sufficient to address rash in most patients, although dose delays may be recommended for more severe cases. However, patients who present with severe rash should be monitored carefully, because SJS and TEN, while rare, are life-threatening toxicities that have occurred after exposure to brentuximab vedotin. In patients who experience these events, treatment should be discontinued and urgent medical care administered.
Infusion-Related Reactions
In the pivotal phase II trials, grade 1 or 2 IRRs were reported in 12% of patients. The most common AEs associated with IRRs were chills, nausea, dyspnea, pruritus, pyrexia, and cough.12Most IRRs reported in the pivotal trials occurred within the first 2 cycles of treatment and correlated with antitherapeutic antibody (ATA) positivity (Seattle Genetics, Inc, unpublished data). Anaphylaxis was reported in 2 patients enrolled in phase I clinical trials, and in both cases, occurred during cycle 2 (Younes et al16and unpublished data). One patient permanently discontinued treatment; the other patient received premedication and was successfully rechallenged in later cycles.
We recommend an evaluation of concomitant medications 2 weeks before initiation of therapy with brentuximab vedotin. In practice, we have observed an increase in IRRs that is specific to fluconazole, an inhibitor of CYP3A4. Discontinuing use of CYP3A4/5 inhibitors and other nonessential medications may decrease the likelihood that the patient experiences an IRR. Patients should be monitored for IRR or anaphylaxis during and after each infusion and should also be informed that symptoms associated with IRRs may occur up to 24 hours after any infusion of brentuximab vedotin. If a patient experiences an IRR, the infusion should be interrupted so that appropriate medical therapy may be instituted. Premedication with acetaminophen, an antihistamine, and a corticosteroid is recommended for subsequent infusions, and at some institutions, is administered to all patients as standard practice. Protocols for successful desensitization to brentuximab vedotin have also been developed and may be considered for patients who continue to experience hypersensitivity reactions.17-19If anaphylaxis occurs, the infusion should be immediately and permanently discontinued and appropriate medical therapy instituted.
Rare Adverse Events Associated With Brentuximab Vedotin
Progressive Multifocal Leukoencephalopathy (PML)
PML is a demyelinating disease of the central nervous system that is caused by opportunistic infection with John Cunningham (JC) virus, a human polyomavirus. In the absence of gold standard criteria, diagnosis is generally based on a combination of clinical, radiographic, and virologic features.20Symptoms progress gradually and may include cognitive and/or speech abnormalities, visual deficits, motor weakness, sensory deficits, loss of coordination, headache, change in personality, or seizures. Patients who are immunosuppressed or have concomitant hematologic malignancies are at risk of developing PML.21PML is estimated to occur in 0.07% to 0.52% of patients with lymphoproliferative disorders who are treated with chemotherapy.21-23PML has been observed in patients treated with brentuximab vedotin in both clinical trials and postmarketing experience at rates of less than 0.1% (TABLE 4). While the frequency of PML in patients treated with brentuximab vedotin falls within the range of background frequencies reported in this patient population, PML represents a serious potential complication that requires appropriate surveillance and patient education about potential symptoms. Dosing with brentuximab vedotin should be suspended if PML is suspected and permanently discontinued if PML is confirmed.
Pancreatitis
Acute pancreatitis is characterized by abdominal pain, nausea, and elevated pancreatic enzymes (serum lipase and/or amylase ≥3 times the upper limit of normal).24Cases of acute pancreatitis in patients who received brentuximab vedotin have been reported in the literature and include both clinical trial and postmarketing experience.25,26The incidence of acute pancreatitis in the clinical trials was 0.25%, and analysis of reports from postmarketing sources yielded an estimated reporting rate of 0.16%. In most cases, patients experienced symptoms within cycle 1 or 2.
The etiology of acute pancreatitis in patients treated with brentuximab vedotin is unclear; many of the cases reported were confounded by underlying diseases or by concomitant medications known to be associated with pancreatitis. In a tissue cross-reactivity study, biotin-conjugated brentuximab vedotin did not demonstrate binding in human pancreas tissues (Seattle Genetics, Inc, unpublished data). It is possible that the hepatic excretion of MMAE in patients with biliary obstruction may lead to pancreatitis. Acute pancreatitis is estimated to occur in 4.4% to 7.7% of patients after hematopoietic stem cell transplant (HSCT).27,28Based on a review of cases reported to date, pancreatitis represents a potential risk after administration of brentuximab vedotin, and appropriate surveillance of symptoms is needed. While routine monitoring of serum amylase and lipase is not recommended, close monitoring for possible pancreatitis, including evaluation of serum lipase and amylase, is recommended for patients who present with severe abdominal pain. In the event that pancreatitis is diagnosed, treatment with brentuximab vedotin should be discontinued and appropriate medical care administered.
Pulmonary Toxicity
In the phase II pivotal trial in HL, pneumonitis was reported as a serious adverse reaction in 2% of patients.12A postmarketing analysis of safety data from patients treated with brentuximab vedotin monotherapy yielded a 0.26% reporting rate of interstitial lung disease (ILD) (TABLE 4). When brentuximab vedotin was studied in combination with ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) or AVD (doxorubicin, vinblastine, and dacarbazine) in a phase I open-label, multicenter, dose-escalation trial, 11 of 25 (44%) patients in the ABVD plus brentuximab vedotin cohort experienced pulmonary toxicity, including 2 patient deaths that were associated with pulmonary toxicity. However, when brentuximab vedotin was combined with AVD in the absence of bleomycin, no pulmonary toxicity was observed.29Because of these findings, concomitant use of bleomycin and brentuximab vedotin is contraindicated.12
A causal association has not been determined for pulmonary toxicity that has been observed with brentuximab vedotin monotherapy. Possible causes include infection and interstitial inflammation. Opportunistic infections should be considered from lymphoma-related immunosuppression. As such, management should include antibiotics, and if the patient is neutropenic, growth factors. Anti-inflammatory agents, including steroids, should be considered if no infectious etiology is identified.
Pulmonary complications following HSCT are reported in 30% to 60% of patients, although this figure is not specific to HL.30In a retrospective review of 184 patients who received ABVD for newly diagnosed HL, bleomycin-induced pneumonitis was experienced by 15% of patients.31Another review of 94 patients with HL who received carmustine-containing high-dose chemotherapy reported a 28% incidence of idiopathic pneumonia syndrome within 1 year of treatment, with 35% mortality.32While the incidence of pulmonary toxicity observed in patients treated with brentuximab vedotin falls within the background frequencies reported in the literature, it remains essential to monitor patients for early symptoms. Particular vigilance should be exercised for patients with one or more risk factors for pulmonary toxicity. Risk factors include advanced age; female gender; prior transplant; graft-vs-host disease; and treatment with high-dose chemotherapy, particularly carmustine.30,31,33Patients should be referred to a specialist if their pulmonary status prohibits them from receiving treatment with brentuximab vedotin.
Hepatotoxicity
Clinical manifestations of drug-induced liver disease vary widely, with acute hepatitis and/or cholestasis being the most common presentations.34Elevated transaminases are the most common sign of hepatotoxicity. The incidence of hepatotoxicity associated with brentuximab vedotin is 1.4% in the clinical trials, and the spontaneous reporting rate from postmarketing sources is 0.61% (TABLE 4). The reports of hepatotoxicity included 17 deaths, of which 5 were considered possibly related to brentuximab vedotin.
The background frequency of hepatotoxicity in patients with lymphoma is unknown. It is estimated that 11% of cases of acute liver failure in the United States may be attributed to idiosyncratic drug-induced liver injury.35Hepatic AEs have also been reported with other microtubule inhibitors, including paclitaxel, docetaxel, and ixabepilone.9,36,37Liver enzymes and bilirubin should be monitored in patients receiving brentuximab vedotin. A lower starting dose of 1.2 mg/kg is recommended for patients who exhibit mild hepatic impairment at baseline, and use of brentuximab vedotin should be avoided in patients with moderate or severe hepatic impairment. Dosing should be decreased or discontinued in patients with new or worsening moderate or severe hepatic dysfunction. However, it is important to distinguish hepatic dysfunction from tumor infiltration of the liver from that due to other causes. We recommend a full dose of 1.8 mg/kg brentuximab vedotin in cases of liver involvement by the patient’s lymphoma, although it must be noted that this recommendation is inconsistent with the prescribing information.
Clinical Pearls
- Toxicities associated with the use of antibody-drug conjugates differ from those associated with naked antibodies because of the presence of cytotoxic agents attached to the antibodies. Brentuximab vedotin consists of a CD30-specific antibody conjugated to monomethyl auristatin E, a microtubule disrupting agent.
- Common adverse events (AEs), such as peripheral neuropathy, associated with brentuximab vedotin may often be managed with modifications to dose and schedule.
- Rare AEs observed in patients treated with brentuximab vedotin, including progressive multifocal leukoencephalopathy, Stevens-Johnson syndrome/toxic epidermal necrolysis, pancreatitis, pulmonary toxicity, and hepatotoxicity, have been reported at rates similar to or below background.
Discussion
Both clinical trial and practical experience suggest that brentuximab vedotin is generally well tolerated, with an AE profile that may be managed by a combination of supportive care and dose modification. Recommendations for surveillance and management of the AEs discussed in this report are summarized inTABLE 5. While the rare AEs discussed here fall within or below the background range reported in the literature in relevant patient populations, vigilant monitoring of symptoms remains crucial to ensuring optimal patient care.
TABLE 5. Summary of Adverse Events and Their Management [CLICK TO DOWNLOAD]
The most commonly reported AE, peripheral neuropathy, is a known class effect of antimicrotubule agents.38,39Preclinical studies have demonstrated the ability of brentuximab vedotin to exert cytotoxic activity on nontarget cells. This bystander effect is mediated by the release of free, membrane-permeable MMAE into the localized microenvironment and may underlie the neuropathy, and potentially other AEs, associated with brentuximab vedotin.4
The correlation between CD30 expression and response to brentuximab vedotin is still being explored. Some patients whose tumor specimens demonstrated very low or undetectable levels of CD30 expression by conventional immunohistochemistry (IHC) have exhibited responses to brentuximab vedotin, but it remains unclear how the effects of the drug are mediated in these patients.13One hypothesis is that CD30 is present and brentuximab vedotin acts through the mechanism described earlier, but levels of CD30 are too low to be detected by routine IHC methods. With the aid of quantitative image analysis, tumor samples previously found to have undetectable levels of CD30 through routine IHC testing have been shown to be CD30 positive.40Some have postulated that the reported cases of toxicities in CD30-negative organs, such as the liver and pancreas, may be attributed to low levels of CD30 in those organs, nonspecific uptake, or may be related to normal metabolic and excretion pathways for MMAE. However, it is also possible that these events are either idiosyncratic in nature or caused by the underlying disease.
The AEs discussed here do not encompass the full spectrum of AEs reported for brentuximab vedotin. Current safety information for approved indications may be found in the prescribing information. Unless otherwise noted, the AEs discussed in this report represent experience with brentuximab vedotin as a single agent. Brentuximab vedotin is not approved for use in combination with other chemotherapeutic agents. Because toxicities associated with combination use are unknown and may vary from those described here, brentuximab vedotin should not be combined with other chemotherapies or with targeted agents unless the combination has proven safe in clinical trials.
Conclusion
ADC technology remains relatively new to oncology treatment, and the AE profiles of ADCs are less fully characterized than those associated with longstanding chemotherapeutic or other biologic treatment options. We hope to establish practical strategies for patient care by presenting our clinical experience with brentuximab vedotin alongside safety information from published clinical trials.
Acknowledgments
The authors wish to acknowledge Ellen Samuelson and Barbara Britt for clinical support, and Olivia Lee for assistance in manuscript preparation. All are employees of Seattle Genetics, Inc.
REFERENCES
- Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30- monomethyl auristatin E conjugate with potent and selective antitumor activity.Blood. 2003;102(4):1458-1465.
- Doronina SO, Toki BE, Torgov MY, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy.Nat Biotechnol. 2003;21 (7):778-784.
- Pettit GR. The dolastatins. Fortschr Chem Org Naturst. 1997;70:1-79.
- Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate.Clin Cancer Res.2010;16(3):888-897.
- Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma.J Clin Oncol.2012;30(18):2183-2189.
- Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study.J Clin Oncol.2012;30(18):2190-2196.
- Hancock SL, Cox RS, McDougall IR. Thyroid diseases after treatment of Hodgkin’s disease.N Engl J Med.1991;325(9):599-605.
- Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy.J Neurol.2002;249(1):9-17.
- IXEMPRA [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2011. http://packageinserts.bms.com/pi/pi_ixempra.pdf. Accessed October 27, 2014.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study.J Neurol Neurosurg Psychiatry. 2000;68(6):750- 755.
- Forero-Torres A, Bartlett NL, Berryman RB, et al. Extended treatment with brentuximab vedotin in patients with relapsed or refractory CD30-positive hematological malignancies [published online October 7, 2014].Leuk Lymphoma.
- ADCETRIS [package insert]. Bothell, WA: Seattle Genetics; 2015: http://www.adcetris.com/pdf/ADCETRIS-brentuximab-vedotin-Prescribing-Information.pdf. Accessed April 13, 2015.
- Jacobsen ED, Advani RH, Oki Y, et al. A phase 2 study of brentuximab vedotin in patients with relapsed or refractory CD30-positive non-Hodgkin lymphomas: interim results.Blood. 2012 2012;120(21): Abstract 2746.
- Advani R, Oki Y, Shustov AR, Grove LE, Bartlett N. Brentuximab vedotin for relapsed or refractory non-Hodgkin lymphoma: Preliminary results from a phase II study.J Clin Oncol.2012 2012;30(suppl 15): Abstract 8070.
- Duvic M, Tetzlaff M, Clos AL, Gangar P, Talpur R. Phase II trial of brentuxi-mab vedotin for CD30+ cutaneous T-cell lymphomas and lymphoproliferative disorders.Blood. 2013;122(21): Abstract 367.
- Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas.N Engl J Med. 2010;363(19):1812- 1821.
- O’Connell AE, Lee JP, Yee C, Kesselheim J, Dioun A. Successful desensiti-zation to brentuximab vedotin after anaphylaxis.Clin Lymphoma Myeloma Leuk. 2014;14(2):e73-e75.
- Story SK, Petrov AA, Geskin LJ. Successful desensitization to brentuximab vedotin after hypersensitivity reaction.J Drugs Dermatol. 2014;13(6):749- 751.
- Devita MD, Evens AM, Rosen ST, Greenberger PA, Petrich AM. Multiple successful desensitizations to brentuximab vedotin: a case report and literature review.J Natl Compr Canc Netw. 2014;12(4):465-471.
- Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section.Neurology. 2013;80(15):1430-1438.
- Bellizzi A, Nardis C, Anzivino E, et al. Human polyomavirus JC reactivation and pathogenetic mechanisms of progressive multifocal leukoencephalopathy and cancer in the era of monoclonal antibody therapies.JNeurovirol.2012;18(1):1-11.
- Power C, Gladden JG, Halliday W, et al. AIDS- and non-AIDS-related PML association with distinct p53 polymorphism.Neurology. 2000;54(3):743- 746.
- Bower JH, Hammack JE, McDonnell SK, Tefferi A. The neurologic complications of B-cell chronic lymphocytic leukemia.Neurology. 1997;48(2):407-412.
- Banks PA, Freeman ML, Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis.Am J Gastroenterol.2006;101(10):2379-2400.
- Urru SA, Mariotti E, Carta P, et al. Acute pancreatitis following brentuximab vedotin therapy for refractory Hodgkin lymphoma: a case report.Drugs R D.2014;14(1):9-11.
- Gandhi MD, Evens AM, Fenske TS, et al. Pancreatitis in patients treated with brentuximab vedotin: a previously unrecognized serious adverse event.Blood. 2014;123(18):2895-2897.
- Shore T, Bow E, Greenberg H, et al. Pancreatitis post-bone marrow transplantation. Bone Marrow Transplant. 1996;17(6):1181-1184.
- Salomone T, Tosi P, Raiti C, et al. Clinical relevance of acute pancreatitis in allogeneic hemopoietic stem cell (bone marrow or peripheral blood) transplants.Dig Dis Sci.1999;44(6):1124-1127.
- Ansell SM, Connors JM, Park SI, O’Meara MM, Younes A. Frontline therapy with brentuximab vedotin combined with ABVD or AVD in patients with newly diagnosed advanced stage Hodgkin lymphoma.Blood. 2012;120(21): Abstract 798.
- Afessa B, Litzow MR, Tefferi A. Bronchiolitis obliterans and other late onset non-infectious pulmonary complications in hematopoietic stem cell transplantation.Bone Marrow Transplant. 2001;28(5):425-434.
- Ngeow J, Tan IB, Kanesvaran R, et al. Prognostic impact of bleomycin-induced pneumonitis on the outcome of Hodgkin’s lymphoma.Ann Hematol.2011;90(1):67-72.
- Rubio C, Hill ME, Milan S, O’Brien ME, Cunningham D. Idiopathic pneumonia syndrome after high-dose chemotherapy for relapsed Hodgkin’s disease.Br J Cancer.1997;75(7):1044-1048.
- Chen YB, Lane AA, Logan BR, et al. Incidence and risk factors for the development of idiopathic pneumonitis syndrome (IPS) after autologous hematopoietic cell transplantation (AutoHCT) for patients with lymphoma.Biol Blood Marrow Transplant. 2014;20(2):S106.
- Kaplowitz N. Drug-induced liver injury.Clin Infect Dis. 2004;38(suppl 2): S44-S48.
- Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury.Mayo Clin Proc. 2014;89(1):95-106.
- TAXOL [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2011. http:// packageinserts.bms.com/pi/pi_taxol.pdf. Accessed October 27, 2014.
- TAXOTERE [package insert]. Bridgewater, NJ: sanofi-aventis U.S. LLC; 2013. http://products.sanofi.us/Taxotere/taxotere.pdf. Accessed October 27, 2014.
- Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents.J Clin Oncol.2006;24(10):1633-1642.
- Swain SM, Arezzo JC. Neuropathy associated with microtubule inhibitors: diagnosis, incidence, and management.Clin Adv Hematol Oncol.2008;6(6): 455-467.
- Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin.Blood. 2014;123(20):3095-3100.















Examining the Non-Hodgkin Lymphoma Treatment Paradigm
July 15th 2022In season 3, episode 6 of Targeted Talks, Yazan Samhouri, MD, discusses the exciting new agents for the treatment of non-Hodgkin lymphoma, the clinical trials that support their use, and hopes for the future of treatment.
Listen



