ATR Inhibitor Combinations Begin to Tackle DNA Damage Repair in Solid Tumors
ATR’s integral role in DNA damage repair has made it an appealing target for cancer treatment, and several ATR inhibitors are being evaluated for efficacy and safety across solid tumors.



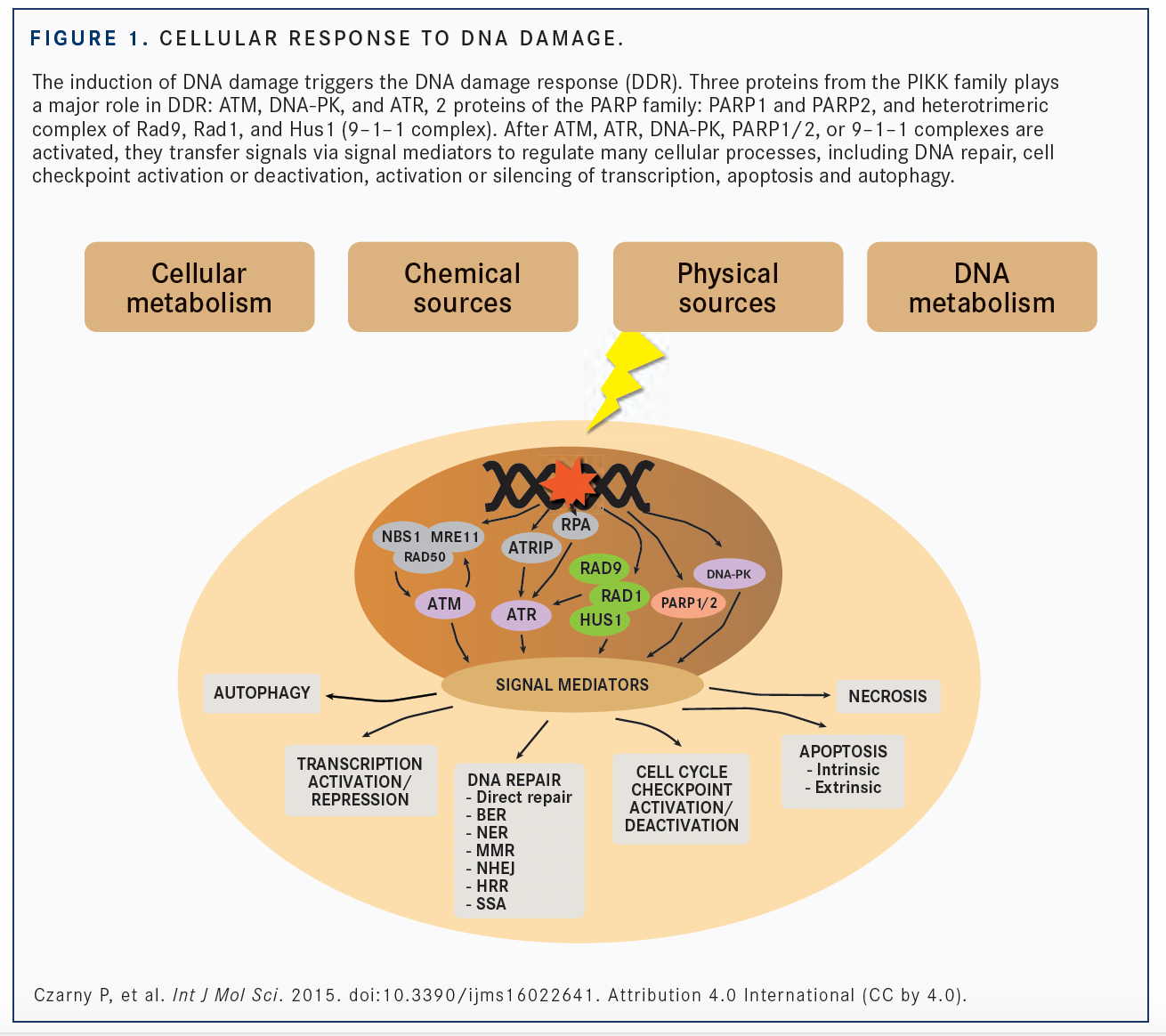
Dysreguation of dysfunction of the DNA damage response (DDR) system can result in genomic instability, a well-known hallmark of many cancers.1,2 DDR deficiency can lead to greater potential for cancer development and malignant transformation (FIGURE), but DDR can also be a mechanism of resistance to chemotherapy and radiotherapy.2 Targeted anti-cancer therapy approaches aim to exploit genetic or functional defects in DDR via synthetic lethal mechanisms with limited effects on healthy tissues.3
PARP is the best-understood component of DDR, and mutations in genes within the Fanconi anemia/BRCA pathway (such as BRCA1, BRCA2) that are common in breast, ovarian, prostate, and pancreatic cancers are most sensitive to PARP inhibitors.3,4 Currently approved PARP inhibitors include olaparib (Lynparza), rucaparib (Rubraca), niraparib (Zejula), and talazoparib (Talzenna).5 However, acquired PARP inhibitor resistance is common.
More targeted therapies aimed at inhibiting DNA damage kinases, classical nonhomologous end joining, and alternative end joining have come from continued DDR research.3 Two regulators of DDR of interest are the DNA damage kinases ataxia telangiectasia mutated Rad3-related kinase (ATR) and its downstream effector checkpoint kinase 1 (CHK1).6 The ATR-CHK1 pathway regulates and coordinates processes such as inhibition of replication origin firing, protection of stressed replication forks, cell cycle arrest, and DNA repair.6 ATR’s contributions to early delay in the M phase entry, regulation of late-phase DDR, and prevention of chromosome breaks during S phase are well described.7
ATR’s integral role in DDR has made it an appealing target for cancer treatment, and several ATR inhibitors are being evaluated or efficacy and safety across solid tumors. Haider S. Mahdi, MD, a gynecologic oncologist at UPMC Hillman Cancer Center at UPMC Magee-Womens Hospital and assistant professor in the Department of Obstetrics, Gynecology, and Reproductive Sciences at the University of Pittsburgh School of Medicine in Pennsylvania, commented in an interview with Targeted Therapies in Oncology™ on the current state of ATR inhibitor therapy: “There are trials looking at patients with solid tumors with deficiency in the DDR pathway; most of them are early-phase trials, and most are cancer specific.”
BAY 1895344: A Potent and Synergistic ATR Inhibitor
BAY 1895344, a novel selective ATR inhibitor, demonstrated high selectivity and potency in a variety of tumor types with DDR defects in initial in vivo and in vitro studies.8 The synthetic lethality of tumor-intrinsic DNA-repair deficiencies and the ATR blockade was confirmed with pronounced in vivo antitumor efficacy of the agent in xenograft models.
Superior antitumor activity as monotherapy when compared with ceralasertib (AZD6738) and berzosertib (M6620; VX-970) was observed; however, BAY 1895344 was most effective in combination with DNA damage-inducing or repair-compromising therapies.8
Synergistic antitumor activity was observed with external beam radiation therapy in colorectal cancer, the PARP inhibitor olaparib in breast and prostate cancers, and with the androgen receptor antagonist darolutamide (Nubeqa) in prostate cancer. However, dose-dependent toxicity was observed in combination with carboplatin, and the combination was less effective than that of BAY 1895344 alone. BAY 1895344 shows promise as a combination therapy, especially with PARP inhibitors and is being evaluated in a phase 1 study in patients with solid tumors and lymphomas (NCT03188965).8
Another study compared castration-resistant prostate cancer (CRPC) cell lines treated with BAY 1895344, olaparib, or BAY 1895344 plus anti–PD-L1. BAY 1895344 destabilized the PD-L1 protein by directly repressing ATR-CHK1 signaling. This resulted in an autocrine, apoptotic response in CRPC cells. Combination of BAY 1895344 with anti–PD-L1 therapy resulted in robust immune activation in the mouse model with a synergistic, T-cell−dependent therapeutic response. Combining ATR-targeted agents with immune checkpoint blockade for patients with CRPC is supported by these mechanisms.9
Ceralasertib in Combination Regimens Ceralasertib + PARP Inhibition
The addition of an ATR inhibitor to a PARP inhibitor is theorized to combat PARP inhibitor resistance. Olaparib was combined with the ATR inhibitor ceralasertib on days 1 to 7 of a 28-day cycle continuously in 25 patients with relapsed or refractory cancers harboring DDR alterations as part of the Olaparib Combinations trial (NCT02576444). Primary end points were confirmed complete response (CR) or partial response (PR) rates and clinical benefit rate (CBR; CR + PR + stable disease [SD] at 16 weeks).10


The overall response rate was 8% and the CBR was 62.5%. The median duration of response was 22 months (range, 18-26+) and the median duration of clinical benefit was 5 months (range, 4-26+).10
ATM (ataxia-telangiectasia mutated) mutations were found in 5 patients, and 2 of these achieved CR or SD ongoing at 24 months or more. Out of 7 patients with BRCA-mutated PARP inhibitor–resistant high-grade serous ovarian cancer, 1 achieved PR (90%) and 5 had SD ranging from 16 to 72 weeks (CBR, 86%). Safety of ceralasertib combined withfull-dose olaparib was confirmed, though dose reduction due to myelosuppression was required in 5 patients. Signs of clinical benefit were observed specifi cally in patients with ATM-deficient cancers and BRCA-mutated, PARP inhibitor–resistant ovarian cancer, encouraging further study.10
The included patients were all heavily pre-treated with a median of 4 prior therapies (range, 0-10) and patients with highgrade serous ovarian cancer were PARP inhibitor–resistant but still saw clinical benefit. “Low response rate could be due to the fact we didn’t limit the previous number of lines of therapy,” commented Mahdi. He noted this study used higher doses of PARP inhibition in comparison to ATR inhibition. “PARP inhibitor–resistant models show ATR is the driver. In this study, we administered a higher dose of PARP with lower dose of ATR inhibition, so maybe we need to focus on the other way around.”
Olaparib is approved for the treatment of /2-mutant, homologous recombination repair (HRR)-deficient pancreatic cancer and was tested in combination with radiation and ceralasertib in tumor models. The combination significantly reduced radiation survival relative to monotherapy of either agent. Therapeutic concentrations of olaparib plus radiation and ceralasertib increased DNA double-strand breaks, and high concentrations of olaparib resulted in ATR-mediated replication stress response antagonized by ceralasertib. Tumor growth was significantly delayed in HRR-proficient tumor xenografts with the regimen. Success of this combination appeared dependent on PARP1-DNA complexes, supporting the development of this combination for tumors resistant to PARP inhibitors.11
Ceralasertib and Chemotherapy
A phase 1 study (NCT02264678) evaluated carboplatin at a fixed dose with escalating doses of ceralasertib in 21-day cycles in patients with advanced solid tumors. The recommended phase 2 dose was 40 mg once daily on days 1 and 2 for ceralasertib with carboplatin at area under the curve 5 every 3 weeks.
Common treatment-emergent adverse events (AEs) observed with the combination were anemia (39%), thrombocytopenia (36%), and neutropenia (25%). Dose-limiting toxicities of grade 4 thrombocytopenia, including a combination of grade 4 thrombocytopenia and grade 3 neutropenia, occurred in 3 of the 36 patients.12 Four patients achieved PR, 2 of which were confirmed and 2 were unconfirmed. Additionally, SD was observed in 18 of 34 (53%) response-evaluable patients. Absent or low ATM or SLFN11 protein expression was found in the 2 patients with confirmed PRs.12
Mahdi commented that platinum-based chemotherapy agents have significant toxicity overlap with ATR inhibition. He stressed that the ATR inhibition dose and/or the chemotherapy dose should be limited because it could be potentially harmful due to increased toxicity. “The potentially more successful approach is to combine it with another agent that is not DNA damaging, such as gemcitabine alone, or [with] the topoisomerase inhibitors, such as topotecan,” he said.
M4344: A Promising ATR Inhibitor
In a preclinical study, the novel ATR inhibitor M4344 was evaluated as monotherapy and in combination with antitumor agents in multiple cancer cell lines, patient-derived tumor organoids, and mouse xenograft models. As a single agent, the potency of M4344 was compared with other investigational ATR inhibitors: BAY 1895344, berzosertib, ceralasertib, and VE-821. M4344 was found to be highly potent; by inducing mitotic catastrophe and DNA damage, its ability to kill cancer cells surpassed that of all the other ATR inhibitors. Additionally, the cytotoxic effects of M4344 were augmented when it was combined with DNA-damaging agents such as topoisomerase inhibitors (eg, topotecan and irinotecan), gemcitabine, cisplatin, and talazoparib; significant synergy was observed. The expression of 2 sets of genes—1 related to replication stress and 1 to neuroendocrine differentiation—was associated with enhanced response to M4344, as was inactivation of the chromatin remodeling genes ARID1A and BRG1. These findings suggest that gene expression signatures and specific genetic tests can be used to predict M4344 activity. Tumors with neuroendocrine characteristics, such as small cell lung cancer and prostate cancer, were significantly correlated with cytotoxic response to M4344. Evaluating these factors may help optimize patient selection for M4344 therapy.13
M4344 demonstrated 100-fold selectivity for ATR over 308 of 312 kinases tested. M4344 suppressed prostate cancer cell proliferation at similar concentrations and similarly to BAY 1895344, but was more potent than berzosertib, ceralasertib, and VE-821. Additionally, M4344 is chemically distinct from other ATR inhibitors. This warrants further studies to determine binding sites and possible interactions between M4344 and ATR.13
Berzosertib: Raising Questions in ATR Inhibition Applications
Berzosertib is an intravenous, highly potent, selective ATR inhibitor. A first-in-human, open-label phase 1 trial (NCT02157792) evaluated the safety and activity of ascending doses of berzosertib in combination with gemcitabine, with or without cisplatin, in patients with advanced solid tumors. Out of 52 patients receiving berzosertib and gemcitabine alone, 4 had a total of 7 dose-limiting toxicities. In the 8 patients receiving additional cisplatin, 3 patients had a total of 3 dose-limiting toxicities.14
Across both cohorts, 5 patients achieved PRs with all 5 having previously received cisplatin or carboplatin. SD was achieved in 29 patients in the berzosertib and gemcitabine cohort plus 4 patients in the triplet cohort.14
Another phase 1 study (NCT02157792) evaluated berzosertib in combination with cisplatin in patients with advanced solid tumors refractory or resistant to standard of care; patients received ascending doses of cisplatin and berzosertib every 3 weeks. A total of 31 patients received berzosertib (90-210 mg/m2) and cisplatin (40-75 mg/m2) across 7 dose levels. PR was observed in 4 patients, all of whom had disease progression following platinum-based chemotherapy, and SD was reached in 15 patients. Neutropenia (20.0%) and anemia (16.7%) were the most common grade 3 or higher treatment-emergent AEs. Two dose-limiting toxicities were observed (grade 3 hypersensitivity and grade 3 increase in alanine aminotransferase).15
A phase 2 study (NCT02567409) evaluated berzosertib in combination with cisplatin and gemcitabine in patients with advanced urothelial carcinoma. A total of 87 patients were randomized to receive cisplatin with gemcitabine plus berzosertib (n = 46) or cisplatin with gemcitabine alone (n = 41).
Median progression-free survival was 8.0 months for both treatment groups. Median overall survival was 14.4 months in the berzosertib group but 19.8 months in patients receiving cisplatin with gemcitabine alone (adjusted HR, 1.42; 95% CI, 0.76-2.68). Higher rates of grade 3 or higher thrombocytopenia (59% vs 39%) and neutropenia (37% vs 27%) were observed with the berzosertib combination vs cisplatin plus gemcitabine alone. AEs within the treatment arm led to necessary dose reductions below the median control arm dose for cisplatin (P < .001).16
“We need to look at specific subsets of cancers and loss-of-function status to determine the functional implications of alteration in the DDR pathway,” Mahdi commented. “It could also be that there is a mutational gene, but the machinery is still intact, so we aren’t seeing expected results.”
Looking Forward
A 2018 pivotal study revealed a mitosisspecific, R loop–driven ATR pathway responsible for suppressing chromosome instability.17 This finding indicates that ATR regulate genomic stability through 2 separate pathways. During S phase, ATR suppresses R loop– associated genomic instability; paradoxically, ATR also mediates the centromeric function of R loops during mitosis.18 Replication stress during S phase and chromosome instability during mitosis are 2 mechanisms of overall genomic instability observed in cancer cells.18,19
Lee Zou, PhD, scientific co-director, Massachusetts General Hospital Cancer Center, and the James & Patricia Poitras Endowed Chair for Cancer Research, Harvard Medical School, Boston, stated, “ATR inhibition is primarily used to target DNA replication stress at this time. There may be opportunities to target mitotic defects with ATR inhibition. Biomarkers predictive of these defects will be important.”
Questions remain regarding ATR inhibition potency, molecular signatures to best predict activity, and most effective therapeutic combinations. ATR kinase and its ATR-CHK1 pathway are essential to DNA integrity, and the clinical success of ATR inhibition, particularly in combination with other DNA-damaging therapies, will require strategies that limit the occurrence of dose-limiting toxicity.
Mahdi indicated that important considerations in ATR inhibition research include, “focusing on higher doses of ATR inhibition and lower doses of PARP inhibition. Either we reverse PARP inhibitor resistance or have another targeted therapy after PARP inhibitor progression. We should also look beyond PARP inhibition or DNA-damaging therapy, combining ATR inhibitors with other agents without overlapping toxicities.”
REFERENCES:
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
2. Tian H, Gao Z, Li H, et al. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015;358(1):8-16. doi:10.1016/j.canlet.2014.12.038
3. Cleary JM, Aguirre AJ, Shapiro GI, D'Andrea AD. Biomarker-guided development of DNA repair inhibitors. Mol Cell. 2020;78(6):1070-1085. doi:10.1016/j.molcel.2020.04.035
4. Gourley C, Balmaña J, Ledermann JA, et al. Moving from poly (adpribose) polymerase inhibition to targeting DNA repair and dna damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257-2269. doi:10.1200/jco.18.02050
5. Chi J, Chung SY, Parakrama R, Fayyaz F, Jose J, Saif MW. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Therap Adv Gastroenterol. 2021;14:17562848211014818. doi:10.1177/17562848211014818
6. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21(21):4780-4785. doi:10.1158/1078-0432.CCR-15-0479
7. Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17(5):615-628. doi:10.1101/gad.1067403
8. Wengner AM, Siemeister G, Lucking U, et al. The novel ATR inhibitor bay 1895344 is effi cacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol Cancer Ther. 2020;19(1):26-38. doi:10.1158/1535-7163.MCT-19-0019
9. Tang Z, Pilie PG, Geng C, et al. ATR Inhibition Induces CDK1-SPOP Signaling and enhances anti-PD-L1 cytotoxicity in prostate cancer. Clin Cancer Res. 2021;27(17):4898-4909. doi:10.1158/1078-0432. CCR-21-1010
10. Mahdi H, Hafez N, Doroshow D, et al. Ceralasertib-Mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (olaparib combinations). JCO Precis Oncol. 2021;5:PO.20.00439. doi:10.1200/po.20.00439
11. Parsels LA, Engelke CG, Parsels J, et al. Combinatorial efficacy of olaparib with radiation and atr inhibitor requires PARP1 protein in homologous recombination-proficient pancreatic cancer. Mol Cancer Ther. 2021;20(2):263-273. doi:10.1158/1535-7163.MCT-20-0365
12. Yap TA, Krebs MG, Postel-Vinay S, et al. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin Cancer Res. doi:10.1158/1078-0432.CCR-21-1032
13. Jo U, Senatorov IS, Zimmermann A, et al. Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol Cancer Ther. 2021;20(8):1431-1441. doi:10.1158/1535- 7163.MCT-20-1026
14. Middleton MR, Dean E, Evans TRJ, et al. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine +/- cisplatin in patients with advanced solid tumours. Br J Cancer. 2021;125(4):510-519. doi:10.1038/s41416-021-01405-x
15. Shapiro GI, Wesolowski R, Devoe C, et al. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br J Cancer. 2021;125(4):520-527. doi:10.1038/s41416-021-01406-w
16. Pal SK, Frankel PH, Mortazavi A, et al. Effect of cisplatin and gemcitabine with or without berzosertib in patients with advanced urothelial carcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 2021;7(10):1536-1543. doi:10.1001/jamaoncol.2021.3441
17. Kabeche L, Nguyen HD, Buisson R, Zou L. A mitosis-specif c and R loop-driven ATR pathway promotes faithful chromosome segregation. Science. 2018;359(6371):108-114. doi:10.1126/science.aan6490
18. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2-9. doi:10.1038/ncb2897
19. Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122(4):1138-1143. doi:10.1172/JCI59954













Gasparetto Explains Rationale for Quadruplet Front Line in Transplant-Ineligible Myeloma
February 22nd 2025In a Community Case Forum in partnership with the North Carolina Oncology Association, Cristina Gasparetto, MD, discussed the CEPHEUS, IMROZ, and BENEFIT trials of treatment for transplant-ineligible newly diagnosed multiple myeloma.
Read More

