To Detect More Alterations, Consider Adding Companion Testing to NGS
Druggable gene aberrations and predictive biomarkers has allowed for the use of next-generation sequencing technologies to grow.


Timothy F. Burns, MD, PhD
Associate Professor of Medicine
Associate Program Director for Research
Hematology/Oncology Fellowship Program
Department of Medicine Division of Hematology-Oncology
University of Pittsburgh Medical Center Hillman Cancer Center
University of Pittsburgh
Pittsburgh, PA

With the emergence of druggable gene aberrations and predictive biomarkers, the use of next-generation sequencing (NGS) technologies has grown. To discern even more characteristics of a tumor, companion diagnostics can provide clinicians with a clearer picture. These diagnostics include the use of RNA sequencing (RNA-seq), which has progressed significantly to enhance transcriptome profiling1 and circulating tumor DNA (ctDNA) detecting.
“Multiple studies over the last 5 to 10 years have shown that using assays that are RNA sequencing–based or adding RNA sequencebased assays to comprehensive next-generation sequencing will help find more targetable fusions,” Timothy F. Burns, MD, PhD, said to Targeted Therapies in Oncology during an interview. Burns is an associate professor of medicine at the University of Pittsburgh, and associate program director for research, Hematology/Oncology Fellowship Program, Department of Medicine at the University of Pittsburgh Medical Center Hillman Cancer Center in Pennsylvania.
The advantages associated with NGS technologies have led them to have distinct advantages over conventional assay techniques, such as single-gene testing, and for expanded targeted mutational platforms in molecular pathology.2 Although conventional assays have high sensitivity and specificity in detecting actionable mutations that match targeted therapies, they also require that each biomarker be prespecified in order to be detected in each purpose-made assay. In contrast, NGS can simultaneously analyze a broad spectrum of genomic alterations that include mutations, translocations, and fusions in multiple genes.2 The result is a more efficient cost analysis when compared to a single biomarker analysis.3
The benefit of enhancing NGS techniques with RNA-seq lies in its ability to provide biological information on bulk RNA seq, to analyze expressions of tumor heterogeneity, and to identify the molecular basis of formation across cancer settings.4 These invaluable insights are important for cancer research and development.
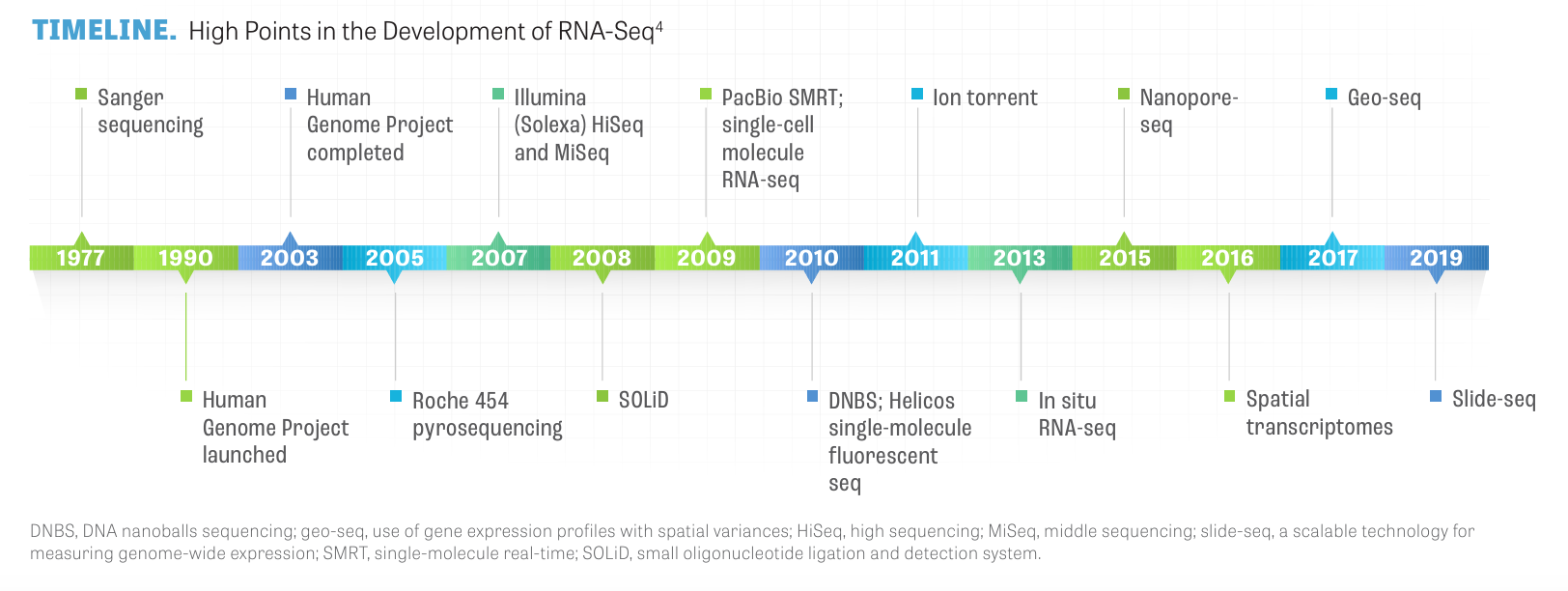
With its ongoing development, RNA-seq has advanced transcriptome analysis from first- generation Sanger sequencing to in situ RNA-seq to slide-seq, as depicted in the TIMELINE.4
Enhanced Transcriptome Analysis
Burns emphasized that the use of RNA-seq is especially useful in lung cancer. “Several FDA approvals for fusion-positive lung cancer, including ALK, ROS1, RET, and NTRK1-3, are now available,” Burns said. “And there are other targetable alterations in addition to fusions, such as MET exon 14 skipping mutations in which an exon is lost. It’s been shown that RNA-seq is better for that [than NGS],” Burns said.
Perhaps RNA-seq’s greatest value is its ability to seek out oncogenic drivers when none might appear initially in NGS. “RNA-seq adds value to NGS. Here [at the UPMC] we have created our own ‘house panel assay,’ ” Burns said. “But in instances when patients are negative for oncogenic drivers, those are the ones that we send out for RNA-seq.” If an oncogenic driver is identified during NGS, it’s likely that a targeted therapy is available or that further testing will reveal a targetable alteration. “If no oncogenic driver is identified, we would keep looking and move on to RNA-seq,” he said.
Gene expression analysis is one of the most common applications for RNA-seq. Samples from different species, tissues, and time periods can be used by RNA-seq to identify differentially expressed genes in order to reveal their function and potential molecular mechanisms. This allows for the discovery of potential cancer biomarkers.3,4
Researchers have noted recurrent gene fusions in breast cancer, including ESR1:: CCDC170, SEC16A::NOTCH1, SEC22B::NOTCH2, and ESR1::YAP1, and, in colorectal cancer, particularly novel configurations of BRAF, NTRK3, and RET gene fusions.5,6


Illustration of DNA double helix
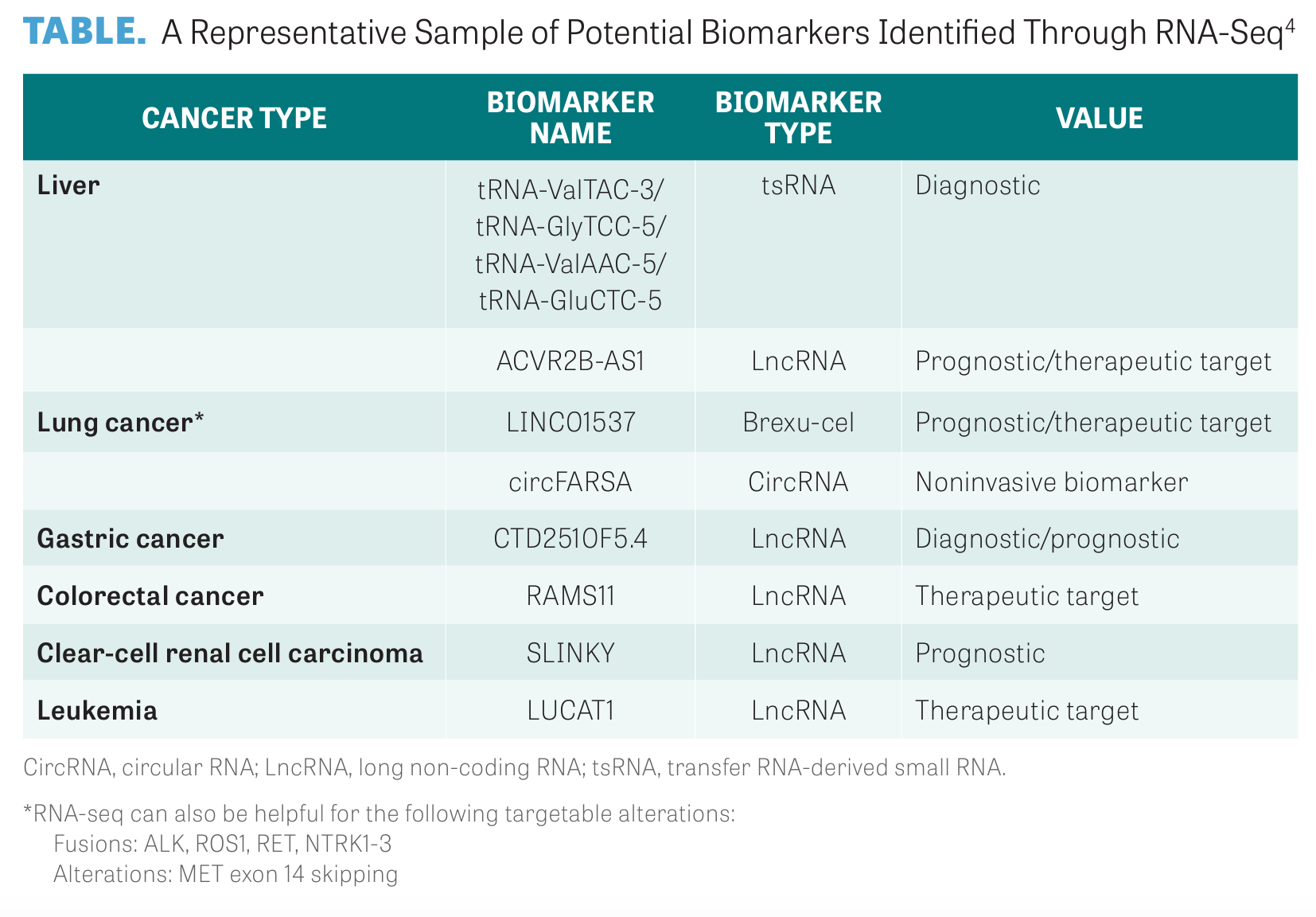
As RNA-seq is further refined, additional differentially expressed genes and biomarkers will be identified, as detailed in the TABLE, but sufficient clinical practice will need to confirm the diagnostic and predictive applications of these biomarkers in cancer.4
The Wait
For all its advancements, sending samples out for NGS and RNA-seq, there remains a time lag to receive the results that averages 2 to 3 weeks, according to Burns. Some cancer and research institutions have developed their own targeted tumor-sequencing test. For example, Memorial Sloan Kettering Cancer Center developed their MSK- IMPACT test, which can detect mutations and other critical changes in the genes of both rare and common cancers.7
The test uses next-generation DNA- sequencing technology that is capable of detecting many classes of genomic changes. These include mutations, gene amplifications and deletions, and genomic rearrangements and signatures such as microsatellite instability and tumor mutation burden.7
“MSK-IMPACT has an extensive panel of genes that are tested for,” Burns said. “To get around the 3-week waiting period, clinicians will run a minimum panel of tests and then run an extended test,” Burns said. “These oncogenic drivers in the treatment-naive setting are mutually exclusive. So if you find one, you don’t usually have to keep looking. For example, if you find a KRAS G12C, KRAS G12D, or KRAS G12V, you’re not going to find an ALK fusion,” Burns said. “And then you’d treat with one of the KRAS inhibitors [depending on the point mutation].”
Immune-Related Adverse Events
Nevertheless, after addressing the patient’s concern and anxiety caused by the delay in results, and providing a way to mitigate tumor burden, clinicians can begin treating with chemotherapy, according to Burns.
“If you don’t know if the patient has an oncogenic driver or not, we can always give a cycle of chemotherapy,” Burns said. “But what’s important to keep in mind is that there are several oncogenic drivers where the therapy is affected when treated by previous immunotherapies. For example, if you have an EGFR or ALK rearrangement and use a targeted inhibitor, the use of prior immunotherapy can affect the rates of adverse events, such as the rates of pneumonitis and hepatitis,” Burns said.8 He warns that it’s possible to compromise a patient’s long-term outcome because they may not be able to now tolerate the targeted therapy.
Drug-related pneumonitis that develops because of the use of immune checkpoint inhibitors is characterized as a clinically significant and potentially life-threatening immune-related adverse event.8
Phase 1 trials of PD-1 inhibitors have resulted in pneumonitis-related deaths in patients with advanced solid tumors, including non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. In a report of long-term safety in an NSCLC cohort that was treated in a phase 1 trial, pneumonitis occurred in 7% (9 of 129) of patients, with 3 pneumonitis- related deaths.9



More data suggest that pneumonitis can be an even more significant issue in patients who are treated with combination therapies. In a phase 1 study of nivolumab (Opdivo) in combination with platinum-based chemotherapy as first-line treatment of advanced NSCLC, pneumonitis was noted in 13% (7 of 56) of patients, including 7% (4 of 56) with grade 3 to 4, and was the most common adverse event responsible for treatment discontinuation (5%; 3 of 56).10
Recently, in a single-institution retrospective study that evaluated 117 patients with primary lung cancer who had received immune checkpoint inhibitors between January 2018 and January 2023, the average time between receiving the first dose of immunotherapy and the development of immunotherapy-induced pneumonitis was 66.5 days (range, 1-163).11
Researchers reported that the average number of cycles received before diagnosis was 3.7, with most cases occurring after fewer than 5 cycles (7 of 10). The grade of pneumonitis ranged from 1 to 5. All patients had NSCLC, with the most prevalent histology being squamous cell carcinoma, ranging from stage III to stage IV.11
“If the patient has a high disease burden, you can always give a cycle of chemotherapy, add immunotherapy after the first cycle,” Burns continued.
If no targetable molecular driver is initially identified and the patient has a PD-L1 expression of 50% or less, those with squamous cell carcinoma are candidates for carboplatin, a taxane, and pembrolizumab, and patients with nonsquamous cell carcinoma are candidates for carboplatin, pemetrexed, and pembrolizumab, or carboplatin, paclitaxel, atezolizumab (Tecentriq), and bevacizumab (Avastin). Patients whose PD-L1 expression is 50% or greater are candidates for pembrolizumab (Keytruda) or nivolumab and ipilimumab (Yervoy).12
ctDNA
Burns said that RNA-seq is an excellent companion test, but it’s not the workhorse diagnostic a lot of community oncologists will be looking to in addition to standard NGS testing. For that, ctDNA offers advantages that have expanded oncologists’ diagnostic tool kit to include the ability to sample bodily fluids such as blood, urine, saliva, cerebrospinal fluid, and pleural effusions.13


“Obtaining ctDNA results is relatively quick, usually 1 week,” Burns said. “For a patient on initial presentation, I’ll do a blood draw for the liquid biopsy and then I’ll schedule their tumor biopsy. When the results from the liquid biopsy come back, I can sometimes cancel the tumor biopsy and treat based on the results of the ctDNA assay,” Burns said. “There are also data suggesting that if you order both a liquid and tumor biopsy, you’ll detect more alterations,” Burns continued. Using liquid biopsy for clinical management, including treatment and disease monitoring, can better represent tumor heterogeneity if multiple tumor types are present, and predictive biomarkers can be successfully detected to guide therapeutic options for NSCLC.14
A question arises, however, when the results of the liquid biopsy come back negative. When this occurs, there could be 1 of 2 explanations: 1) it could be a true negative and the sample revealed no alterations; or 2) it could be a false negative because there wasn’t enough tumor DNA to be detected, or the sample had enough tumor DNA, and the test gave you back alterations but it wasn’t sensitive enough to detect a targetable fusion. “Essentially, if nothing is detected by a liquid biopsy, you probably need to keep looking, and that’s where the tumor biopsy comes in,” Burns said.


On the Horizon
Looking beyond ctDNA, Burns is excited by the prospects of what will be available in terms of next steps. “Response to therapy, especially in the early-stage setting, will be particularly exciting,” Burns said. In particular, measurable residual disease (MRD) assays, which can be used after therapy or after resection when there is no molecularly detectable disease, represent an opportunity to intervene at that point. “I think that’s what’s next—that is, moving beyond just diagnosis but using MRD to guide therapy,” Burns said.
Simone E. Dekker, MD, PhD, and colleagues, explored the use of MRD in acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and chronic myeloid leukemia (CML).15
They note that in AML, MRD is evolving toward use as a clinical decision support tool, but standardization of the technology across laboratories and among technologies needs international efforts. MRD has a major impact on clinical decision-making in patients with ALL. For example, MRD status influences treatment regimens and the decision to proceed with allogeneic hematopoietic cell transplantation. Emerging data suggest that more sensitive modalities that use NGS are superior in predicting outcomes in patients with ALL. And in CML, MRD is well standardized and fully established to guide treatment decision-making during treatment, and before and after stopping tyrosine kinase inhibitors.15
REFERENCES:
1. Hong M, Tao S, Zhang L, et al. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol. 2020;13(1):166. doi:10.1186/s13045-020-01005-x
2. Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020;12(1):8. doi:10.1186/s13073-019-0703-1
3. Moorcraft SY, Gonzalez D, Walker BA. Understanding next generation sequencing in oncology: a guide for oncologists. Crit Rev Oncol Hematol. 2015;96(3):463-474. doi:10.1016/j. critrevonc.2015.06.007
4. Govindarajan M, Wohlmuth C, Waas M, Bernardini MQ, Kislinger T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J Hematol Oncol. 2020;13(1):134. doi:10.1186/s13045-020-00971-6
5. Veeraraghavan J, Ma J, Hu Y, Wang XS. Recurrent and pathological gene fusions in breast cancer: current advances in genomic discovery and clinical implications. Breast Cancer Res Treat. 2016;158(2):219-232. doi:10.1007/ s10549-016-3876-y
6. Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017;77(14):3814-3822. doi:10.1158/0008-5472.CAN-16-3563
7. MSK-IMPACT: a targeted test for mutations in both rare and common cancers. Memorial Sloan Kettering Cancer Center. Accessed March 10, 2024. https://tinyurl. com/46edtnxm
8. Nishino M, Hatabu H, Hodi FS, Ramaiya NH. Drug-related pneumonitis in the era of precision cancer therapy. JCO Precis Oncol. 2017;1:PO.17.00026. doi:10.1200/PO.17.00026
9. Gettinger SN, Horn L, Gandhi L, et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33(18):2004-2012. doi:10.1200/JCO.2014.58.3708
10. Rizvi NA, Hellmann MD, Brahmer JR, et al. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(25):2969-2979. doi:10.1200/ JCO.2016.66.9861
11. Winter M, Gilani MA, Anipindi M, Venigalla T, Doreswamy S. Timing of immunotherapy-induced pneumonitis in patients with primary lung malignancy. J Clin Pathology. 2023;41(suppl 16):2647. doi:10.1200/JCO.2023.41.16_suppl.2647
12. NCCN. Clinical Practice Guidelines in Oncology. Non-small cell lung cancer, version 2.2024 Accessed March 10, 2024. https://www.nccn.org/professionals/physician_gls/ pdf/nscl.pdf
13. Bao Y, Zhang D, Guo H, Ma W. Beyond blood: advancing the frontiers of liquid biopsy in oncology and personalized medicine. Cancer Sci. Published online February 3, 2024. doi:10.1111/cas.16097
14. Casagrande GMS, Silva MO, Reis RM, Leal LF. Liquid biopsy for lung cancer: up-to-date and perspectives for screening programs. Int J Mol Sci. 2023;24(3):2505. doi:10.3390/ ijms24032505
15. Dekker SE, Rea D, Cayuela JM, Arnhardt I, Leonard J, Heuser M. Using measurable residual disease to optimize management of AML, ALL, and chronic myeloid leukemia. Am Soc Clin Oncol Educ Book. 2023;43:e390010. doi:10.1200/EDBK_390010




















Survivorship Care Promotes Evidence-Based Approaches for Quality of Life and Beyond
March 21st 2025Frank J. Penedo, PhD, explains the challenges of survivorship care for patients with cancer and how he implements programs to support patients’ emotional, physical, and practical needs.
Read More



