Integrating Mutational Analysis Into the Clinical Management of Patients With Myelofibrosis
Myelofibrosis is a myeloproliferative neoplasm characterized by splenomegaly, progressive cytopenias, and transformation to acute myeloid leukemia. This review will describe mutations detected in myelofibrosis and discuss how to incorporate mutation information into risk stratification and therapeutic decision making for patients with myelofibrosis.
John Mascarenhas, MD
Abstract
Myelofibrosis is a myeloproliferative neoplasm characterized by splenomegaly, progressive cytopenias, and transformation to acute myeloid leukemia. Several somatic mutations are pathogenetically responsible for this phenotype, the most important of which are JAK2, CALR, and MPL. However, the advent of high-throughput next-generation sequencing has identified multiple other molecular alterations that hold prognostic and possibly therapeutic potential. This tool is now commercially available, yet clinicians are frequently unfamiliar with how to interpret the results and incorporate them into the care of an individual patient. This review will describe mutations detected in myelofibrosis and discuss how to incorporate mutation information into risk stratification and therapeutic decision making for patients with myelofibrosis.
Introduction
Myelofibrosis (MF) is a myeloproliferative neoplasm (MPN) characterized by progressive cytopenias, splenomegaly, bone marrow fibrosis, and clonal proliferation of myeloid cells. MF can be either primary (PMF) or secondary, arising from antecedent essential thrombocythemia (ET) or polycythemia vera (PV), termed PET/PPV-MF. The natural progression of MF is to bone marrow failure and then evolution to acute myeloid leukemia (AML), which portends a bleak prognosis.1Overall survival (OS) is influenced by a number of clinical variables, including advanced age, anemia, red blood cell transfusion dependence, thrombocytopenia, presence of peripheral blood blasts, and karyotypic abnormalities. These risk factors are encapsulated in currently utilized risk stratification tools, such as the Dynamic International Prognostic Scoring System (DIPSS) and DIPSS Plus.2
High-throughput next-generation sequencing (NGS) to detect the presence of acquired somatic mutations holds the potential to not only enhance predictive abilities of clinical scoring systems, but also to personalize therapeutic approaches with mutation-directed targeted therapy.3Technologic advancements in DNA sequencing have decreased the turnaround time while reducing costs, allowing for widespread commercial availability and expanding the application in routine clinical care.4Therefore, it is important for clinicians to be familiar with the clinical applications of NGS in prognostication and therapeutic decision making when caring for patients with MF.
We will first describe the most commonly mutated genes in MF and their prevalence. A thorough review of their prognostic potential including the integration of genomic data into current risk stratification tools will then be presented. Then we will describe the current utilization of mutational analysis in determining the treatment plan and discuss the development of molecularly based targeted therapy. Finally, we advocate for routine integration of mutational profiling into routine clinical practice.
In MF, mutations involving 3 genesJAK2, CALR,and MPL—are known to be directly related to the MPN phenotype. Janus-associated kinase 2 (JAK2) encodes a tyrosine kinase integral to hematopoietic cell function. The gain-of-function JAK2 V617F results in the constitutive activation of the JAK-signal transduction and activator of transcription (STAT) signaling pathway and culminates in upregulation of downstream targets.5Additionally, JAK2 V716F has been shown to directly phosphorylate histone H3, resulting in epigenetic transcriptional changes including upregulation expression of the hematopoietic oncogene LMO2.6Calreticulin (CALR) encodes for a calcium-binding protein, which localizes to the endoplasmic reticulum and has a multitude of intracellular, extracellular, and cell-surface functions. This protein was first discovered to play a role in protein homeostasis but subsequently has been found to be involved in immune regulation, calcium metabolism, phagocytosis, cell adhesion, and migration.7-9CALR frameshift mutations within exon 9 result in activation of the JAK/STAT signaling pathway,10and interact with the thrombopoietin receptor, encoded by MPL, causing overactivation of the JAK/STAT pathway.11 Several recurrent mutations exist in MPL, the most common being MPLW515L/K in which the tryptophan between the cytosolic and transmembrane domains is altered, leading to dimerization of the MPL transmembrane helix and constitutive activation of JAK signaling.12The commonality among these mutations is that they all result in JAK/STAT pathway activation, the central pathobiologic mechanism of MF.13


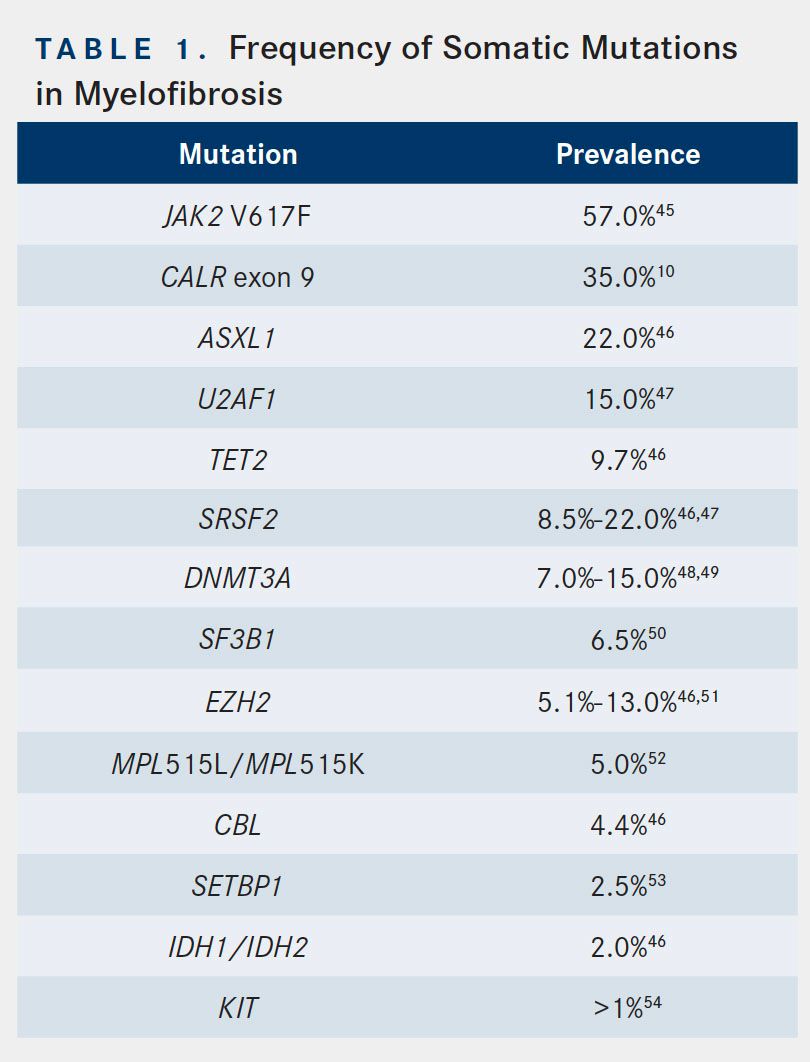
Additionally, other acquired subclonal mutations are present in MF. The prevalence of these mutations is noted in Table 1. Many of these mutations occur in genes that are important for epigenetic regulation of gene expression. Some examples are ASXL1, EZH2, TET2, and DNMT3A, with ASXL1 mutations being the most prevalent and most associated with adverse outcomes across the myeloid malignancies.14,15ASXL1 encodes a scaffolding protein involved in epigenetic regulation,16and its mutations promote myeloid transformation via loss of polycomb repressive complex 2mediated histone H3 lysine 27 methylation.17 Another subset of genes found mutated in MF are those involved in mRNA splicing, including SRSF2 and SF3B1.18Although less prevalent than the aforementioned mutations, IDH1/IDH2 mutations are of particular interest in light of the development of isocitrate dehydrogenase (IDH) inhibitors currently in trial and, in the case of IDH2, now approved. IDH1/IDH2 encode IDH, which catalyzes oxidative decarboxylation of isocitrate to alpha-ketoglutarate. The resultant decrease in alpha-ketoglutarate and/or accumulation of 2-hydroxyglutarate is believed to be oncogenic.19Notably, IDH1/IDH2 mutations are found in a significant percentage of patients with MPN, especially those whose disease transforms to AML; however, these mutations are very rare in patients with MPN in chronic phase.20
Risk Stratification
Several prognostic scoring systems have been developed and employed to risk-stratify patients with PMF. The International Working Group for Myelofibrosis Research and Treatment devised the International Prognosis Scoring System (IPSS) based on a retrospective cohort study of 1054 patients with PMF from 7 centers. Five factors were noted in multivariate analysis to hold independent prognostic significance: presence of constitutional symptoms (ie, weight loss >10%, night sweats, fever), age >65 years, hemoglobin <10 g/dL, leukocyte count >25,000/microL, and circulating blast cells ≥1%.21DIPSS includes the same 5 factors but assigns 2 points for anemia.22 Both the IPSS and DIPSS are calculated by adding points weighted by their corresponding hazard ratio to calculate 4 discrete risk categories of low, intermediate-1, intermediate-2, and high risk. The DIPSS is routinely used in clinical practice to determine a prognostic category at any time during the MF clinical course. Given a growing understanding of clinical features that also influence outcomes independent of the established DIPSS, this scoring system was again updated to include red blood cell transfusion dependence, thrombocytopenia, and unfavorable karyotype, each receiving an additional point. This enhanced prognostication score, DIPSS Plus, was validated in 793 patients with PMF at Mayo Clinic, where the median overall survival with 0 points (low risk), 1 point (intermediate-1), 2 to 3 points (intermediate-2), or 4 to 6 points (high risk) was 15.4, 6.5, 2.9, and 1.3 years, respectively.2
These prediction models may be enhanced by incorporating data from mutational profiling, as this technology can delineate prognostically distinct molecular subtypes. In particular, knowledge about the driver mutational status in patients with MF is essential when counseling patients on their prognosis and in guiding treatment decisions. This is best demonstrated by a study of 428 patients with MF in which the median overall survival in JAK2-, MPL-, and CALR-mutated patients was 5.9, 9.9, and 15.9 years, respectively. Importantly, in the absence of all 3 driver mutations designated as “triple negative” status (TN)—the median survival was only 2.3 years; TN status portends the worst prognosis. In terms of leukemic transformation, TN carries the highest risk, and CALR-mutated in the absence of other mutations carries the most favorable risk.23Interestingly, type 1 (52 base-pair deletions) CALR mutations may be associated with a longer survival versus type 2 (5 base-pair insertions), according to results of a study of 358 patients with PMF. However, in multivariable analysis, which included DIPSS Plus score and ASXL1 mutational status, this survival difference disappeared.24
Aside from the main driver mutations, other somatic mutations also hold significant prognostic information. In a study of 879 patients with MF initiated in a European cohort and validated in a Mayo Clinic cohort, mutations involving ASXL1, SRSF2, and EZH2 were found to be associated with a shorter OS. However, only mutated ASXL1 remained independent of DIPSS, effectively identifying this mutation as a negative prognostic factor with potential to enhance established risk stratification systems. There was discrepancy between cohorts on the mutations found to be associated with leukemic transformation and shortened leukemiafree survival (LFS); however, the results of the study indicated that IDH1/IDH2, SRSF2, EZH2, and ASXLI mutations portend a poor prognosis. A separate study of 254 patients with MF shed further light on molecular-based risk stratification. Specifically, TN status was associated with a median overall survival of 2.5 years; however, patients harboring an ASXL1 mutation without a CALR mutation carried the worst prognosis, with a median overall survival of 2.3 years.25Together, this identifies TN status and ASXL1-mutated/CALR wild-type as high-risk molecular subtypes of MF.
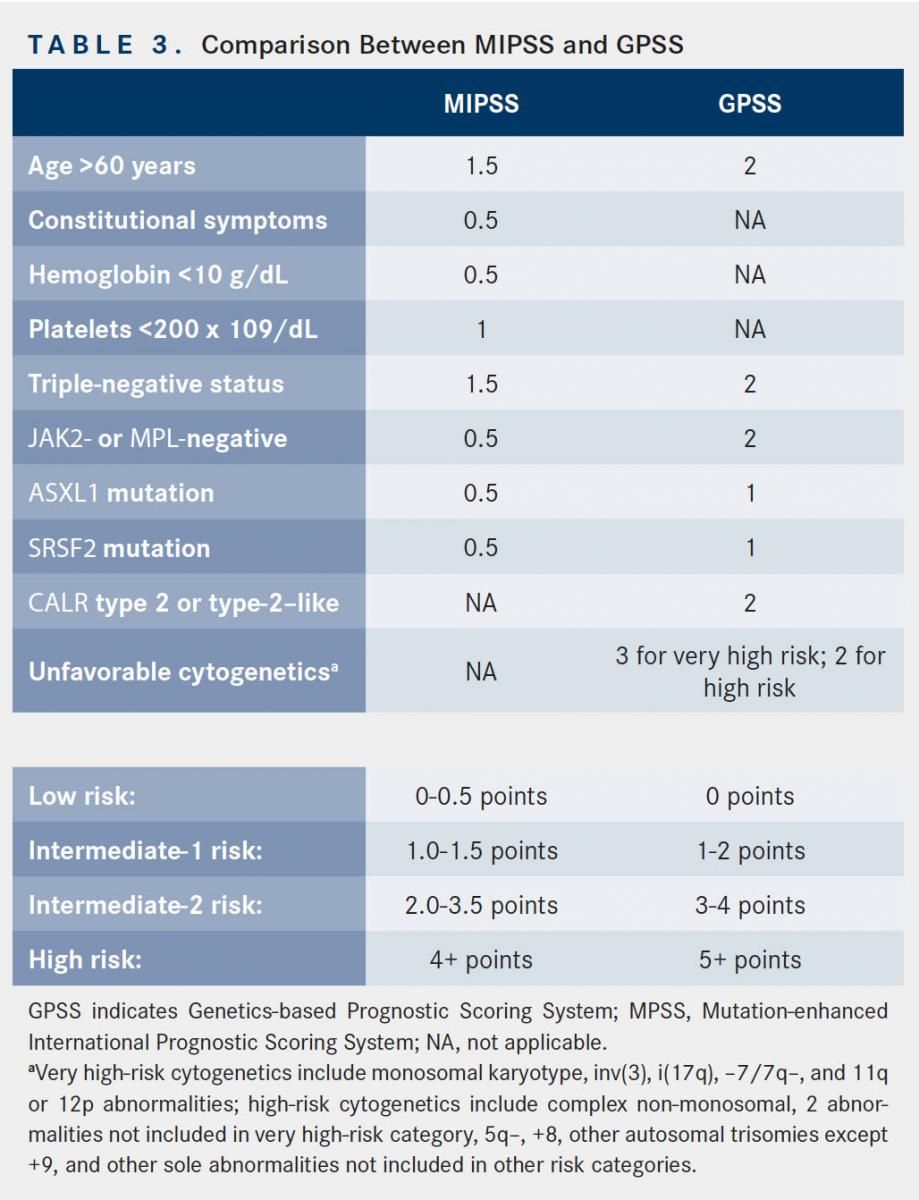
The number of high-risk mutations (ie, ASXL1, EZH2, SRSF2, and IDH1/IDH2) may also be important, as demonstrated in a study of 797 patients with PMF. The cohort with 2 or more high-risk mutations had a median OS of 2.6 years, compared with 7.0 years and 12.3 years, respectively, in the groups with 1 and 0 high-risk mutations. The presence of 2 or more mutations was also associated with a shorter LFS when compared with patients with no prognostically detrimental mutations.26Other mutations may also share prognostic importance in MF. In a series including 197 patients with PV, ET, or PMF, the presence of mutated TP53 and TET2 were independently associated with shorter OS and shorter LFS.27 However, other studies have noted a neutral effect of TET2 on survival and leukemic transformation.28Table 2 summarizes the current prognostic knowledge of these mutations in MF. Recently, several risk stratification tools have been developed that incorporate mutational information to refine prognostication and therefore improve treatment decision making. One such scoring system is the mutation-enhanced IPSS (MIPSS), which takes into account the mutational status of CALR, JAK2, and MPL; TN status; each single variable included in the IPSS; and additional key detrimental mutations (ASXL1, SRSF2, EZH2, and IDH1/IDH2) to create 4 distinct risk categories (Table 3). MIPSS performed better than IPSS in predicting survival (1611.6vs 1649.0) based on Akaike information criterion, which estimates the quality of a statistical model.29A second novel prognostic model, the Genetic-based Prognostic Scoring System, takes into account age, karyotyping, and mutational information.30However, the utility of these scoring systems in routine clinical practice remains unknown as they have not been prospectively validated.



Impact on Treatment
Treatment with ruxolitinib (Jakafi), a JAK1/JAK2 inhibitor, is the sole FDA-approved treatment for patients with MF. Ruxolitinib significantly reduces splenomegaly and improves symptom burden in patients with MF.31Importantly, this clinical benefit accrues regardless of JAK2 mutational status or the presence of prognostically detrimental mutations (ASXL1, EZH2, SRSF2, IDH1/IDH2).32 However, the number of mutations present may predict spleen response with ruxolitinib treatment. This finding was noted in a posthoc analysis of 95 patients in a phase I/II trial of ruxolitinib. The number of mutations present was inversely correlated with spleen response; patients with ≤2 mutations had 9-fold higher odds of spleen response compared with those with ≥3 mutations. Additionally, patients with 3 or more of these high-risk mutations (ASXL1, EZH2, IDH1, or IDH2) had a shorter time to treatment discontinuation and shorter OS.33 Thus, the absence of JAK2 V617F does not preclude a patient from benefit with ruxolitinib treatment. However, high mutational burden (ie, more than 3 mutations) likely represents more aggressive disease and portends a worse outcome even with the use of ruxolitinib. NGS may therefore be a useful tool for prognostication as well as in the context of discussing the role of ruxolitinib versus consideration of clinical trial or hematopoietic stem cell transplantation (HSCT).
With the expansion of mutational profiling utilizing NGS panels, novel targeted therapies are actively being evaluated in MF. Particular attention has been focused on the inhibition of IDH2, as IDH2 mutations are associated with more advanced forms of MF, including MPN blast phase.34A specific potent reversible inhibitor of mutant IDH2, enasidenib (Idhifa), has been shown in multiple models to dramatically reduce the level of 2HG in acute myeloid leukemia (AML) cells, supporting enasidenib’s clinical development.35In vitro use of these inhibitors, in addition to the first-in-human phase I/II study of enasidenib, have shown proof-of-concept and promising data that such targeted therapy can produce cytostatic effects and induce terminal cellular differentiation in patients with mutant-IDH2 relapsed/ refractory AML.36,37Based on the favorable clinical response rates, the FDA approved enasidenib for the treatment of relapsed or refractory AML harboring an IDH2 mutation.38Given the prevalence and association of IDH2 mutations with blast-phase MPN, there is potential for these molecularly targeted novel agents to prevent or treat leukemic transformation, as well as to treat secondary AML arising from MPN. Additionally, preclinical evidence in a JAK2/IDH2 co-mutated murine model suggests that combined JAK2 and IDH2 inhibition decreases evidence of disease burden and reverses abnormal gene expression in hematopoietic stem cells to a greater extent than JAK2 inhibition alone.39


There are a paucity of data on how mutational status affects HSCT outcomes. In patients with low or intermediate-1 risk by DIPSS, the risk of death outweighs the potential benefit of HSCT. However, in eligible patients with intermediate-2 or high-risk disease, there is potential benefit.40Some experts have also reasoned that patients with high-risk mutations, such as ASXL1 or SRSF2, or TN disease, should proceed to HSCT earlier than patients without these mutations regardless of their clinical risk score.20,21The role of driver mutations JAK2, MPL, and CALR in HSCT remains unclear. There may, however, be a potential benefit of molecular genotyping to predict outcomes after HSCT. Several studies exemplify this potential role in the HSCT setting. One study of 133 patients with PMF receiving HSCT found that patients with mutated CALR fared better than patients with wild-type CALR post transplant. When compared with their wild-type counterparts, the patients with mutated CALR demonstrated better 4-year OS, better nonrelapse mortality (NRM), and a trend toward improved cumulative incidence of relapse. Overall, patients with mutated CALR were found to have the best prognosis, patients with JAK2 or MPL were found to have intermediate prognosis, and patients with TN status clearly had the worst prognosis.41The association of these mutational statuses with posttransplant prognosis parallels the findings in nontransplant patients, with CALR mutants faring best, patients with TN status faring worst, and those harboring mutated JAK2 and MPL having intermediate prognosis.25
Another study looked specifically at the role of the JAK2 V617 in 162 patients with MF undergoing HSCT. Patients with JAK2 wild-type demonstrated decreased OS after HSCT when compared with patients harboring the JAK2 V617F mutation. However, achieving JAK2 V617F negativity by highly sensitive real-time polymerase chain reaction assay after HSCT was associated with a decrease in relapse.42These results have been duplicated and expanded to include nondriver mutations including ASXL1, SRSF2, EZH2, and IDH1/IDH2, in a retrospective study of 169 patients with MF. Patients with CALR mutations were found to have lower NRM and improved PFS and OS, while ASXL1 and IDH2 mutations were associated with lower PFS. TN status and SRSF2 or EZH2 mutations were not associated with poorer outcome post HSCT.35The small number of patients studied in each group does, however, limit the generalizability of the findings.43
Further validation is required in large prospective studies, but the results of these studies exemplify the potential for genomic data to refine risk stratification. It is evident that the presence of a poor prognostic mutation in a patient categorized as intermediate-1 or low-risk by DIPSS category likely warrants more aggressive treatment than the same DIPSS risk patient without such a high-risk mutation. To formalize this logic, it is imperative to adapt risk stratification to include genomic mutations.
There has also been an attempt to investigate the role of early treatment with ruxolitinib in patients with MF with low-risk disease but high-risk mutations, using a clinical scoring system. The ReTHINK trial was a multicenter, double-blind, placebocontrolled, phase III study that set out to accrue 320 patients with MF who had a low symptom burden; nonpalpable or barely palpable spleen; and at least 1 of the following mutations: ASXL1, EZH2, SRSF2, and IDH1/IDH2.44 However, this trial was recently terminated because of poor accrual (NCT02598297).
Conclusions
Mutational profiling has revolutionized oncologic care. In the case of MF, NGS has the potential to aid in prognostication through refined risk stratification and thereby assist in guiding treatment decisions for individual patients. Furthermore, similar to developments in the fields of other related myeloid malignancies and in oncology in general, movement toward molecular-based personalized therapies will become commonplace. This will drastically change the treatment paradigm for a molecularly heterogeneous population of patients. Genomic-based technology is commercially available and should be integrated into the routine care of patients with MF. As biotechnology and molecular insights increase in MF, it is imperative that clinicians continue to stay abreast of these advances to provide state-of-the-art care for their patients.
References:
- Mesa RA, Li CY, Ketterling RP, et al. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973-977.
- Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. doi: 10.1200/JCO.2010.32.2446.
- Rampal R, Levine RL. A primer on genomic and epigenomic alterations in the myeloproliferative neoplasms. Best Pract Res Clin Haematol. 2014;27(2):83-93. doi: 10.1016/j.beha.2014.07.001.
- Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila). 2012;5(7):887-900. doi: 10.1158/1940- 6207.CAPR-11-0432.
- Levine RL, Loriaux M, Huntly BJ, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106(10):3377-3379.
- Dawson MA, Bannister AJ, Göttgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819-822. doi: 10.1038/nature08448.
- Michalak M, Groenendyk J, Szabo E, et al. Calreticulin, a multi-process calciumbuffering chaperone of the endoplasmic reticulum. Biochem J. 2009;417(3):651- 666. doi: 10.1042/BJ20081847.
- Imai M, Araki M, Komatsu N. Somatic mutations of calreticulin in myeloproliferative neoplasms. Int J Hematol. 2017;105(6):743-747. doi: 10.1007/ s12185-017-2246-9.
- Gold LI, Eggleton P, Sweetwyne MT, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24(3):665-683. doi: 10.1096/ fj.09-145482.
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390. doi: 10.1056/NEJMoa1311347.
- Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPLdependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317-1324. doi: 10.1182/blood-2015-11-679571.
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667-679. doi: 10.1182/blood-2016-10-695940.
- Rumi E, Pietra D, Pascutto C, et al; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062-1069. doi: 10.1182/blood-2014-05-578435.
- Carbuccia N, Murati A, Trouplin V, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183-2186. doi: 10.1038/ leu.2009.141.
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1-2):21-29. doi: 10.1016/j. mrfmmm.2008.07.010.
- Katoh M. Functional and cancer genomics of ASXL family members. Br J Cancer. 2013;109(2):299-306. doi: 10.1038/bjc.2013.281.
- Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180-193. doi: 10.1016/j.ccr.2012.06.032.
- Lehmann U, Bartels S, Hasemeier B, et al. SRSF2 mutation is present in the hypercellular and prefibrotic stage of primary myelofibrosis. Blood. 2013;121(19):4011-4012. doi: 10.1182/blood-2012-11-467662.
- Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261-265. doi: 10.1126/science.1170944.
- Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122-2126. doi: 10.1182/blood-2009-11-250878.
- Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901. doi: 10.1182/blood-2008-07-170449.
- Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703-1708. doi: 10.1182/blood-2009-09-245837.
- Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507-2513; quiz 2615. doi: 10.1182/blood-2014-05-579136.
- Tefferi A, Lasho TL, Finke C, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28(7):1568-1570. doi: 10.1038/leu.2014.83.
- Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triplenegative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28(7):1472-1477. doi: 10.1038/leu.2014.3.
- Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804-1810. doi: 10.1038/ leu.2014.76.
- Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220- 2228. doi: 10.1182/blood-2013-11-537167.
- Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23(5):905-911. doi: 10.1038/leu.2009.47.
- Vannucchi AM, Guglielmelli P, Rotunno G, et al. Mutation-enhanced International Prognostic Scoring System (MIPSS) for primary myelofibrosis: an AGIMM & IWG-MRT project. Blood. 2014;124(21):405-405. bloodjournal.org/ content/124/21/405?sso-checked=true.
- Tefferi A, Guglielmelli P, Finke C, et al. Integration of mutations and karyotype towards a Genetics-based Prognostic Scoring System (GPSS) for primary myelofibrosis. Blood. 2014;124(21):406-406. bloodjournal.org/ content/124/21/406.
- Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-798. doi: 10.1056/NEJMoa1110556.
- Guglielmelli P, Biamonte F, Rotunno G, et al; COMFORT-II Investigators; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) Investigators. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. 2014;123(14):2157-2160. doi: 10.1182/blood-2013-11-536557.
- Patel KP, Newberry KJ, Luthra R, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126(6):790-797. doi: 10.1182/blood-2015-03-633404.
- Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302-1309. doi: 10.1038/leu.2010.113.
- Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478-493. doi: 10.1158/2159-8290.CD-16-1034.
- Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622-626. doi: 10.1126/science.1234769.
- Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi: 10.1182/ blood-2017-04-779405.
- FDA granted regular approval to enasidenib for the treatment of relapsed or refractory AML [news release]. Silver Spring, MD: FDA; August 1, 2017. fda.gov/Drugs/ InformationOnDrugs/ApprovedDrugs/ucm569482.htm. Accessed April 6, 2018.
- McKenney AS, Lau AN, Somasundara AVH, et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J Clin Invest. 2018;128(2):789-804. doi: 10.1172/JCI94516.
- Keyzner A, Han S, Shapiro S, et al. Outcome of allogeneic hematopoietic stem cell transplantation for patients with chronic and advanced phase myelofibrosis. Biol Blood Marrow Transplant. 2016;22(12):2180-2186. doi: 10.1016/j. bbmt.2016.08.029.
- Panagiota V, Thol F, Markus B, et al. Prognostic effect of calreticulin mutations in patients with myelofibrosis after allogeneic hematopoietic stem cell transplantation. Leukemia. 2014;28(7):1552-1555. doi: 10.1038/leu.2014.66.
- Alchalby H, Badbaran A, Zabelina T, et al. Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood. 2010;116(18):3572-3581. doi: 10.1182/ blood-2009-12-260588.
- Kroger N, Panagiota V, Badbaran A, et al. Impact of molecular genetics on outcome in myelofibrosis patients after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(7):1095-1101. doi: 10.1016/j.bbmt.2017.03.034.
- Passamonti F, Kiladjian J-J, Vannucchi AM, et al. ReTHINK: a randomized, doubleblind, placebo-controlled, multicenter, phase 3 study of ruxolitinib in early myelofibrosis patients. J Clin Oncol. 2016;34(suppl 15):TPS7080-TPS7080. doi: 10.1200/JCO.2016.34.15_suppl.TPS7080
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
- Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861-1869. doi: 10.1038/leu.2013.119.
- Martinez-Avilés L, Besses C, Alvarez-Larrán A, et al. Mutations in the RNA splicing machinery genes in myelofibrotic transformation of essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2014;164(4):605-607. doi: 10.1111/bjh.12647.
- Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217-1219. doi: 10.1038/ leu.2011.77.
- Abdel-Wahab O, Pardanani A, Rampal R, et al. DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leukemia. 2011;25(7):1219-1220. doi: 10.1038/ leu.2011.82
- Lasho TL, Finke CM, Hanson CA, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia. 2012;26(5):1135-1137. doi: 10.1038/leu.2011.320.
- Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722- 726. doi: 10.1038/ng.621.
- Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472-3476.
- Laborde RR, Patnaik MM, Lasho TL, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10):2100-2102. doi: 10.1038/ leu.2013.97.
- Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428- 2436. doi: 10.1200/JCO.2012.47.3314.














Survivorship Care Promotes Evidence-Based Approaches for Quality of Life and Beyond
March 21st 2025Frank J. Penedo, PhD, explains the challenges of survivorship care for patients with cancer and how he implements programs to support patients’ emotional, physical, and practical needs.
Read More






