Evolving Paradigms in Immuno-Oncology: Introduction and Overview
This feature covers the "Introduction" and "Overview of Mechanisms" sections of the current Evolving Paradigms in Immuno-Oncology issue.
Introduction
The activation of the immune system to combat cancerous tumor cells through immune checkpoint inhibition, rather than targeting the tumor directly, has become a strong focus of research and clinical development in recent years. Naturally, the immune system is involved in tumor inhibition through immune surveillance, identifying cancerous or precancerous cells based on antigens expressed in the context of cellular stress, resulting in activation of cytotoxic T cells to destroy these cancerous cells before they can cause harm and multiply.1Within this process, immune checkpoints function to control the balance of costimulatory and regulatory signals essential to maintaining self-tolerance, as well as regulating amplitude and duration of T-cell response. Recently, these checkpoints have offered new targets for therapeutic intervention in oncology, allowing for reinvigoration of the adaptive immune system and native antitumor response. Specifically, the use of antibodies targeting the cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed death receptor-1 (PD-1), and its ligand, PD-L1, have demonstrated impressive results for the treatment of several historically difficult-to-treat tumor types, ushering in multiple new therapeutic options.
Overview and Mechanisms of Checkpoint Inhibition
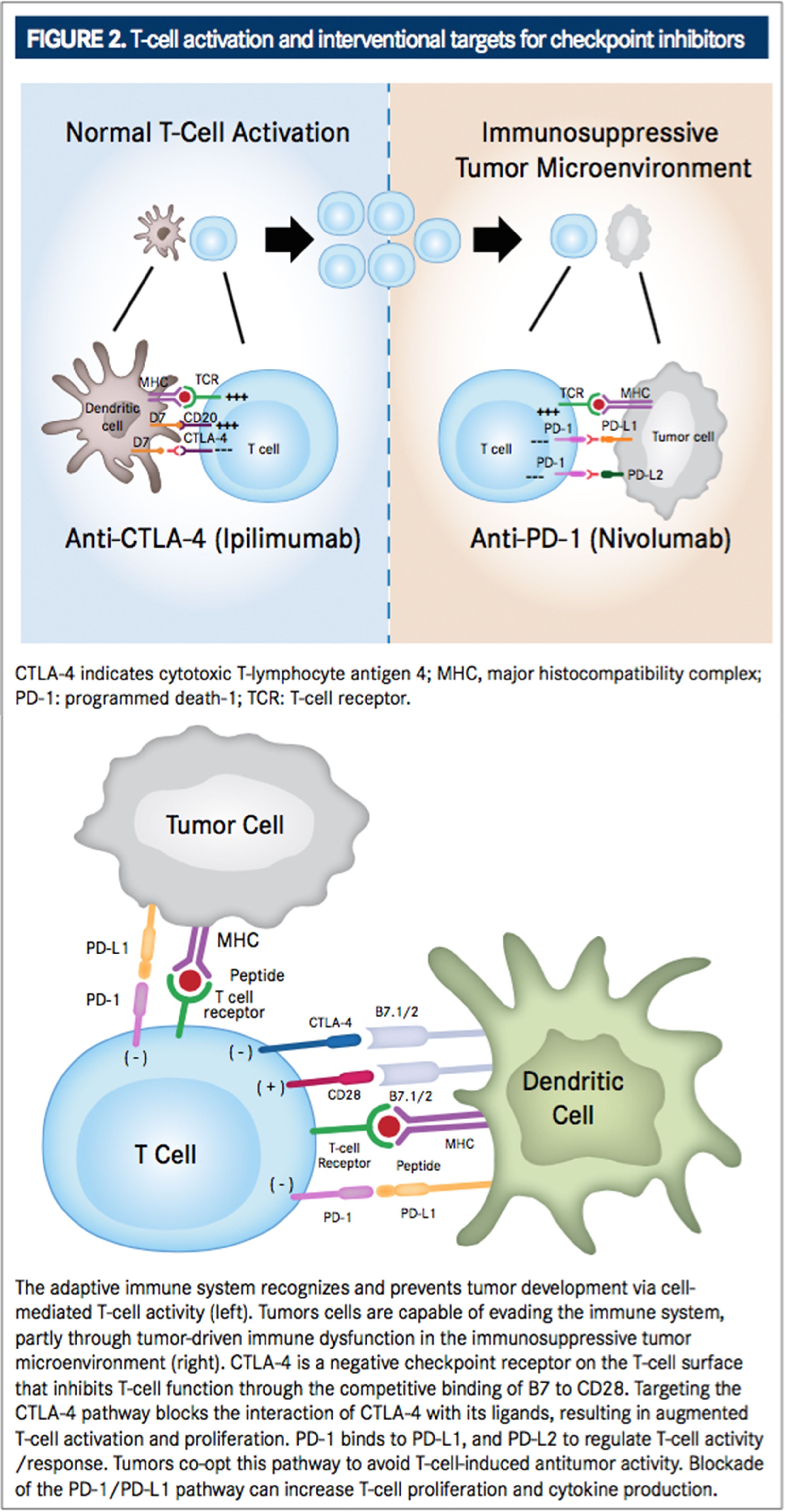
The adaptive immune system recognizes and prevents tumor development via cell-mediated T-cell activity, particularly through a subset of cytotoxic T lymphocytes (CTLs), which express cell surface-associated alpha-beta T-cell receptors (TCRs) and the co-receptor CD8+. Yet, tumors still develop in the presence of a healthy immune system, indicating cancer cells are capable of evading the immune system, partly through tumor-driven immune dysfunction.2Immunotherapies provoke a biological immune response toward cancer cells, stimulate immunologic memory, and enable a patient’s own immune system to attack cancerous cells in a self-sustained manner with the intent of preventing tumorigenesis long after therapy.
Immunotherapy was first applied to oncology in 1881 when William Coley, a bone sarcoma surgeon, administered streptococcal organisms (or other bacteria/bacterial products), now termed “Coley’s Toxins,” to patients. This marked the first recorded study of a systemic immunotherapy in the treatment of malignant tumors.3In 1898, Coley published a paper noting spontaneous remission in patients receiving a series of killed infectious agents (ie, Coley’s Toxins).3Fifty years later (1951), further immunotherapeutic attempts were made by Higgins and Pack who tested virus inoculation in 30 patients with malignant melanoma, observing a reduction in size of lesions in six patients.4Then, in the late 1960s, the study of immunotherapies in patients with advanced melanoma was furthered; a bacilli Calmette-Guérin (BCG) vaccine (an attenuated preparation of cultured Mycrobacterium bovis that produces a nonspecific immuno-boosting response) was shown to be efficacious in small clinical trials.5Further investigation of BCG led to its approval for the treatment of recurrent localized bladder cancer in 1989.6During the late 20th century, several other key aspects of immune regulation emerged, leading to the approval of the first modern immunotherapies. In 1986, interferon (IFN)alpha 2A and 2B were approved for the treatment of hairy cell leukemia.7By 1991, improvements in response rates for patients with metastatic melanoma were identified with the use of IFNs in addition to dicarbazine chemotherapy (DTIC).8This was followed by the approval of aldesleukin (Proleukin, Prometheus), a recombinant interleukin (IL)-2, for the treatment of metastatic renal cell carcinoma (mRCC) in 1992.8-10Then, in the late 1990s, research was published demonstrating improvements in complete and partial response rates among patients with stage III and IV melanoma receiving a high-dose interleukin (IL)-2 regimen.11Research further evolved with the improved understanding of immune checkpoint inhibition, leading to the testing of urelumab (anti-CD137), the first agonistic checkpoint inhibitor to enter clinical studies with two phase I studies in 2008.8Unfortunately the program was halted due to the occurrence of excessive liver toxicities, but the fundamental approach motivated the development of other immunotherapies employing a similar concept of immune checkpoint inhibition.8


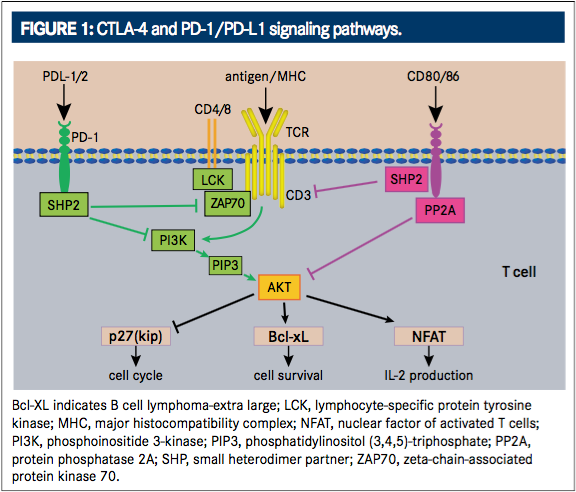
At the turn of the 21st century, understanding of the CTLA-4 pathway resulted in a great breakthrough for immunotherapy treatment. The CTLA-4 pathway was first identified as a potential target for anticancer therapy in 2001 when in vivo blockade of the CTLA4 receptor revealed antitumor effects mediated by regulation of Tlymphocyte proliferation.12During T-cell activation in a normally functioning immune system, an antigen-independent costimulatory signal is produced when B7 molecules (CD80 and CD86, also called B7-1 and B7-2, respectively) on the antigen presenting cell surface bind with CD28 receptors on the T-ell surface (FIGURE 1).13This signal elicits the expression of CTLA-4. CTLA-4 is a negative checkpoint receptor on the T-cell surface that inhibits T-cell function through the competitive binding of B7 to CD28 by interacting with B7 ligands (CD80 and CD86) at a much higher affinity than CD28. This negative checkpoint prevents subsequent costimulatory signals and dampens T-cell activation and proliferation in order to prevent autoimmune reactions against healthy tissue while the immune system is activated against a pathogen.8,14,15Targeting the CTLA-4 pathway (FIGURE 2) blocks the interaction of CTLA-4 with its ligands resulting in augmented T-cell activation and proliferation, including activation and proliferation of tumor-infiltrating Teffector cells. Further, blockade of CTLA-4 also reduces T-regulatory cell function, which contributes to an overall increase in T-cell responsiveness, including antitumor immunity.16In 2010, the safety, efficacy, and utilization of such a therapy became actualized after demonstration of improved overall survival (OS) benefit for patients with metastatic melanoma in a phase III, randomized controlled trial. This resulted in the 2011 FDA approval of ipilimumab (Yervoy, Bristol-Myers Squibb) for the treatment of unresectable or metastatic melanoma, making it the first checkpoint inhibitor approved for the treatment of cancer.16-18Ipilimumab is currently the only approved CTLA-4 inhibitor for the treatment of cancer, although a second antiCTLA-4 receptor antibody, tremelimumab, is in clinical development. It was recently reported by its manufacturer (AstraZeneca) to have received orphan drug designation for the treatment of malignant mesothelioma.14


While CTLA-4 expression is focused on regulating the activation of T cells, a second checkpoint receptor that regulates effector T cell activity in peripheral tissues, notably in response to infection or tumor progression, has been identified and targeted in tumor treatment.19PD-1 is a transmembrane inhibitory protein and member of the CD28 receptor family that is not detected on resting T cells, but is highly expressed on activated T cells. Its role in immune regulation has been known for decades,8as it was first isolated by subtractive-hybridization in 1992 as a molecule whose expression is enhanced by apoptotic stimuli.20It shares 21% to 33% amino acid homology with CTLA-4/CD28/inducible co-stimulator (ICOS; a molecule on activated T cells that has been shown to deliver a stimulatory co-signal to other activated T cells upon engagement of its ligand, ICOS-L.).20Similar to ICOS, but in contrast to CD28 and CTLA-4, PD-1 is expressed on T cells upon activation, as well as on B cells and monocytes.20Also, although both CTLA-4 and PD-1 inhibit serine threonine kinase AKT (also known as protein kinase VB), PD-1 does so through its effects on the phosphoinositide 3-kinase (PI3K) pathway, whereas CTLA-4 effectively inhibits AKT independent of PI3K (FIGURE 1).2
PD-1 and its ligands, PD-L1 (previously referred to as B7 homolog 1 [B1-H1] or CD274) and PD-L2 (previously referred to as B7 dendritic cell [B7-DC]) play an important role in regulation of T-cell activity through inhibition of T-cell proliferation and cytokine production by activated T cells. This helps to maintain equilibrium between T-cell activation and tolerance in prevention of autoimmune damage. PD-L1 has thus been implicated in the promotion of tumor growth.13,21,22The binding of PD-1 to its ligands can inhibit cytotoxic T-cell response, and signaling through this pathway can impede the active T-cellmediated immune surveillance of tumors.23,24 To this end, tumor-infiltrating CD8+ T cells with elevated expression of PD-1 have been correlated with impaired T-cell function. As such, blockade of the PD-1/PD-L1 pathway (FIGURE 2) can increase T-cell proliferation and cytokine production.13Tumors have seemingly co-opted the PD-1/PD-L1 pathway to avoid T-cellinduced antitumor activity, as PD-L1 is highly expressed in many types of tumors and PD-L2 is found to be upregulated in some tumors.21,25,26For example, CD8+ T cells from patients with NSCLC have been observed to display substantially elevated PD-1 expression, and expression of PD-L1 and PD-L2 has been demonstrated on various tumor cells lines. PD-L1 expression on tumor cells (both in vitro and in vivo) is associated with tumor escape and greater tumorigenesis/tumor persistence.20,27
Further, PD-L1 and PD-L2 differ in their expression patterns.28PD-L1 has been shown to be constitutively expressed and upregulated on murine hematopoietic cells (eg, T cells, B cells, macrophages, DCs, and bone marrow-derived mast cells) as well as on nonhematopoietic cells (eg, endothelial, epithelial, and muscle cells), whereas PD-L2 is only inducibly expressed on DCs, macrophages, and bone marrow-derived mast cells.28In addition, PD-L1 expression is potently induced by IFN-gamma as well as several other proinflammatory molecules, including tumor necrosis factor (TNF)-alpha, lipopolysaccharide (LPS), granulocyte-macrophage colony-stimulating factor (GM-CSF), and vascular endothelial growth factor (VEGF). Activated T cells produce high levels of IFN-γ and other cytokines, creating a highly immunosuppressive tumor microenvironment.29,30Further, the engagement of PD-L1 or PD-L2 with PD-1 delivers an inhibitory co-signal, suggesting that PD-1 has multiple roles in immune regulation.20Thus, PD-L1 makes an optimal target for immunotherapy in oncology.
In the last few months of 2014, two new monoclonal antibodies (mAbs) targeting the PD-1 receptor were approved: pembrolizumab (Keytruda, Merck) and nivolumab (Opdivo, Bristol-Myers Squibb). These PD-1 inhibitors decrease tumor growth by binding to the PD-1 receptor and blocking its interaction with PD-L1 and PD-L2, releasing PD-1 pathway inhibition of the immune response, including the antitumor immune response.23,24Both pembrolizumab and nivolumab were initially approved for the treatment of unresectable or advanced melanoma, with approval later extended to nonsmall cell lung cancer (NSCLC) and in the case of nivolumab, RCC, all under accelerated review as a second-line treatment for patients with progression following chemotherapy in NSCLC or following antiangiogenic therapy in RCC.23,24Additionally, two therapeutic inhibitors of PD-L1 are currently in late-phase clinical trials for cancer treatment: durvalumab (MEDI4736; AstraZeneca) and atezolizumab (MPDL3280A; Genentech). Both are high-affinity engineered human antiPD-L1 IgG1 mAbs that block PD-L1 from binding to its receptors, PD-1 and B7-1 (CD80).31,32
Combination therapy with both CTLA-4 and PD-1 blockade therapies has also emerged as a prospective treatment option providing synergistic antitumor treatment. Although CTLA-4 and PD-1 are both inhibitory receptors and checkpoints in T cell’s activation, the pathways are nonredundant, fulfill distinct roles, and mediate their effects through separate mechanisms. For example, the intracellular pathways through which each receptor transmits signals are different (eg, PD-1 inhibition of the serine threonine kinase Akt activation via its effects on the PI3K pathway, while CTLA-4 inhibits Akt independent of PI3K).2
Interestingly, the CTLA-4 ligand CD80 (B7-1) has also been identified to bind to PD-L1, but not PD-L2, and it has been established that signals through both B7-1 and PD-L1 are inhibitory. The interaction of B7-1 and PD-L1 result in diminished expression of cell-surface activation markers, decreased T-cell proliferation, and reduced cytokine production.28,33This is significant because it means that B7-1 has binding sights for both PD-L1 and CTLA-4 and that PD-L1 has both B7-1 and PD-1 binding sights.28 Additionally, “reverse signaling” has been observed via PD-L1 and PD-L2 molecules expressed on dendritic cells that can either enhance or inhibit dendritic cell activation (measured by DC maturation and cytokine production).2













Key Trials From ASH 2024 Impact Treatment for Plasma Cell Disorders Going Forward
February 20th 2025Peers & Perspectives in Oncology editorial board member Marc J. Braunstein, MD, PhD, FACP, discussed the significant advancements in multiple myeloma treatment at the 2024 ASH Annual Meeting and Exposition.
Read More

